一种亚苄基樟脑磺酸的制备方法与流程
本发明涉及一种安全性高、环保问题少的亚苄基樟脑磺酸制备方法。
背景技术:
专利US 4323549中example 8和example 9介绍的亚苄基樟脑磺酸(3-benzylidene-2-oxo-10-bornane sulfonic acid)制备方法,以樟脑磺酸和苯甲醛为原料,于甲苯中在甲醇钠的作用下生成亚苄基樟脑磺酸钠,再于水中用盐酸转化成亚苄基樟脑磺酸。该专利方法工艺操作繁琐,原料成本高,废水量大,溶剂采用易制毒品甲苯,并且从甲苯中分离亚苄基樟脑磺酸钠也有一定的危险性。
技术实现要素:
本发明针对现有技术存在的问题,提供一种亚苄基樟脑磺酸的制备方法,降低了原料成本,减少了废水排放,安全性高。
为此,本发明采用的技术方案是:一种亚苄基樟脑磺酸的制备方法,其特征在于是采用苯甲醛和樟脑磺酸为原料,在相转移催化剂和碱的作用下,生成亚苄基樟脑磺酸钠,再用酸调节PH=1,使得亚苄基樟脑磺酸于水中析出,再通过抽滤分离出亚苄基樟脑磺酸。
作为优选,所述的相转移催化剂为四丁基溴化铵、吡啶、18冠6、环糊精、PEG-400、15冠5。
作为优选,所述的碱为氢氧化钠、氢氧化钾、氢氧化锂、碳酸钾、碳酸钠。
作为优选,所述的酸为盐酸、硫酸、磷酸、醋酸。
作为优选,所述的相转移催化剂为PEG-400,所述的碱为30%氢氧化钠,所述的酸为硫酸。
作为优选,所述的苯甲醛是通过滴加的方法进行添加。
作为优选,所述的苯甲醛滴毕后,保温搅拌2h,之后搅拌控温至50℃,用酸缓慢调节PH=1后,搅拌降温至≤10℃并控温≤10℃保温搅拌1h。
作为优选,整个制备过程是在10-100℃下进行。
作为优选,制备过程中滤液可套用多次,套用过程中不需要再次额外添加相转移催化剂,除第一次套用外,滤液套用需要用活性炭脱色,套用的滤液投入反应瓶后于45℃,-0.1MPa条件下进行浓缩,后加入100g樟脑磺酸,搅拌至基本溶清再加入碱进行PH调节。
作为优选,在相转移催化剂和碱的作用下调节PH至7-8。
本发明和现有技术相比,所涉及的亚苄基樟脑磺酸制备方法原料成本低,废水量少,安全性高,适用于工业化生产。
附图说明
图1是本发明实施例1的HPLC谱图。
图2是本发明实施例2的HPLC谱图。
图3是本发明实施例3的HPLC谱图。
图4是本发明实施例4的HPLC谱图。
图5是本发明实施例5的HPLC谱图。
具体实施方式
下面对结合附图对本发明作进一步描述:
实施例1
将100g樟脑磺酸和100g水投入500ml四口瓶,于25-30℃搅拌溶清。搅拌下往四口瓶加入58g 30%氢氧化钠,调节PH=7-8。如果此时体系不溶清,补加适量水确保体系呈溶清状态。在体系溶清的前提下,往四口瓶加入5g PEG-400。搅拌升温至45℃。
控温45±2℃,往四口瓶缓慢滴加45g苯甲醛,约5h滴毕。滴毕控温45±2℃保温搅拌2h。
搅拌升温至50℃,用50g 50%硫酸缓慢调节PH=1后搅拌降温至≤10℃并控温≤10℃保温搅拌1h。
抽滤,滤饼用适量水洗涤,得155g亮白色结晶性固体粉末湿品和淡黄色滤液。
将所得湿品于80-90℃减压干燥至恒重,得130g亮白色结晶性固体粉末干品,每分子该产品含一分子结晶水。
用国食药监许[2010]456号附件中所述的亚苄基樟脑磺酸检测方法检测实施例1所得产品,具体结果如下:
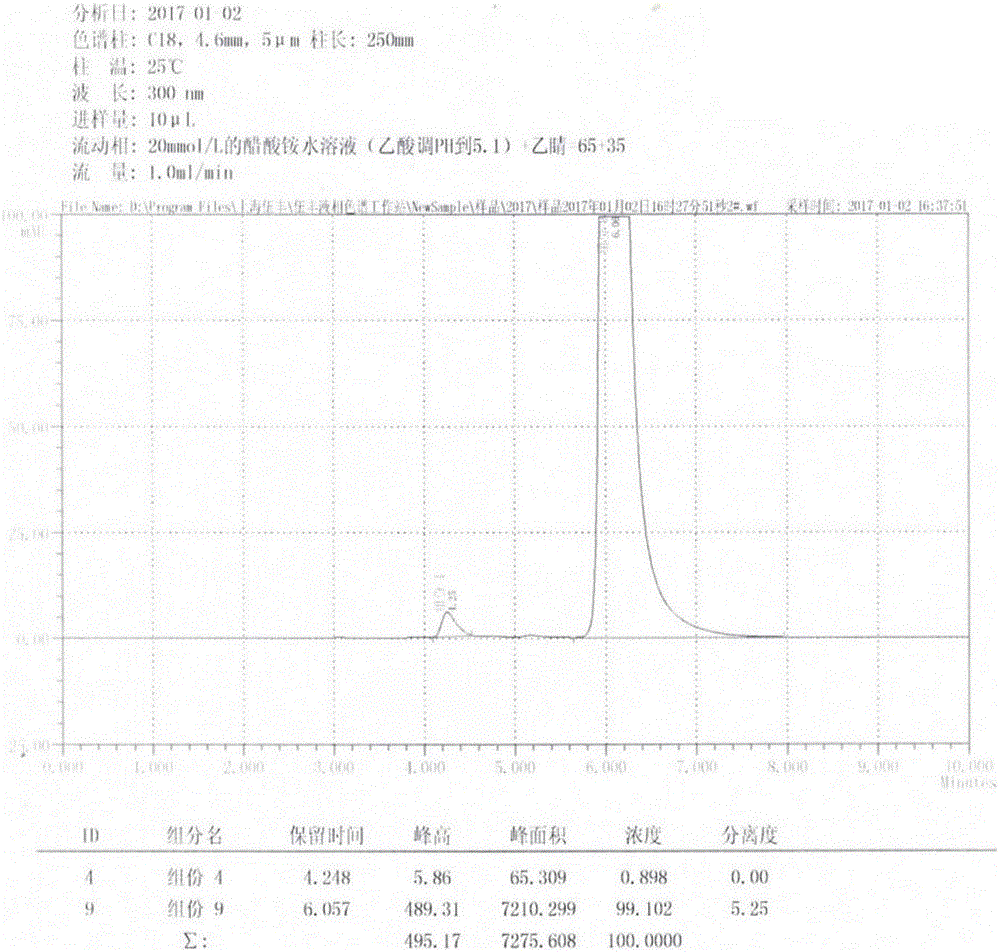
HPLC条件:
色谱柱:C18柱,250mm×4.6mm,5μm;
流动相:20mmol/L的醋酸铵水溶液(乙酸调PH=5.1)+乙腈=65+35;
检测波长:300nm;
柱温:25℃;
进样量:10μL;
样品处理:50mg(精确到0.01mg)于10mL量瓶中,加入硫酸甲醇溶液溶解并定容至10mL。
HPLC谱图如图1所示。
实施例2
将100g樟脑磺酸和100g水投入500ml四口瓶,于25-30℃搅拌溶清。搅拌下往四口瓶加入58g 30%氢氧化钠,调节PH=7-8。如果此时体系不溶清,补加适量水确保体系呈溶清状态。在体系溶清的前提下,往四口瓶加入5g 18冠6。搅拌升温至45℃。
控温45±2℃,往四口瓶缓慢滴加45g苯甲醛,约5h滴毕。滴毕控温45±2℃保温搅拌2h。
搅拌升温至50℃,用51g 50%硫酸缓慢调节PH=1后搅拌降温至≤10℃并控温≤10℃保温搅拌1h。
抽滤,滤饼用适量水洗涤,得132g亮白色结晶性固体粉末湿品和淡黄色滤液。
将所得湿品于80-90℃减压干燥至恒重,得111g亮白色结晶性固体粉末干品,每分子该产品含一分子结晶水。
用国食药监许[2010]456号附件中所述的亚苄基樟脑磺酸检测方法检测实施例2所得产品,具体结果如下:
HPLC条件:
色谱柱:C18柱,250mm×4.6mm,5μm;
流动相:20mmol/L的醋酸铵水溶液(乙酸调PH=5.1)+乙腈=65+35;
检测波长:300nm;
柱温:25℃;
进样量:10μL;
样品处理:50mg(精确到0.01mg)于10mL量瓶中,加入硫酸甲醇溶液溶解并定容至10mL。
HPLC谱图如图2所示。
实施例3
将100g樟脑磺酸和100g水投入500ml四口瓶,于25-30℃搅拌溶清。搅拌下往四口瓶加入58g 30%氢氧化钠,调节PH=7-8。如果此时体系不溶清,补加适量水确保体系呈溶清状态。在体系溶清的前提下,往四口瓶加入5g PEG-400。搅拌升温至60℃。
控温60±2℃,往四口瓶缓慢滴加45g苯甲醛,约5h滴毕。滴毕控温60±2℃保温搅拌2h。
搅拌降温至50℃,用50g 50%硫酸缓慢调节PH=1后搅拌降温至≤10℃并控温≤10℃保温搅拌1h。
抽滤,滤饼用适量水洗涤,得107g浅棕色结晶性固体粉末湿品和淡黄色滤液。
将所得湿品于80-90℃减压干燥至恒重,得91g浅棕色结晶性固体粉末干品,每分子该产品含一分子结晶水。
用国食药监许[2010]456号附件中所述的亚苄基樟脑磺酸检测方法检测实施例3所得产品,具体结果如下:
HPLC条件:
色谱柱:C18柱,250mm×4.6mm,5μm;
流动相:20mmol/L的醋酸铵水溶液(乙酸调PH=5.1)+乙腈=65+35;
检测波长:300nm;
柱温:25℃;
进样量:10μL;
样品处理:50mg(精确到0.01mg)于10mL量瓶中,加入硫酸甲醇溶液溶解并定容至10mL。
HPLC谱图如图3所示。
实施例4
将实施例1中的滤液投入500ml四口瓶,于45℃,-0.1MPa条件下浓缩至120g左右,加入100g樟脑磺酸,搅拌至基本溶清。搅拌下往四口瓶加入62g 30%氢氧化钠,调节PH=7-8。此时体系有少许不溶物,抽滤,澄清的淡黄色滤液搅拌升温至45℃。
控温45±2℃,往四口瓶缓慢滴加45g苯甲醛,约5h滴毕。滴毕控温45±2℃保温搅拌2h。
搅拌升温至50℃,用56g 50%硫酸缓慢调节PH=1后搅拌降温至≤10℃并控温≤10℃保温搅拌1h。
抽滤,滤饼用适量水洗涤,得161g亮白色结晶性固体粉末湿品和淡黄色滤液。
将所得湿品于80-90℃减压干燥至恒重,得139g亮白色结晶性固体粉末干品,每分子该产品含一分子结晶水。
用国食药监许[2010]456号附件中所述的亚苄基樟脑磺酸检测方法检测实施例4所得产品,具体结果如下:
HPLC条件:
色谱柱:C18柱,250mm×4.6mm,5μm;
流动相:20mmol/L的醋酸铵水溶液(乙酸调PH=5.1)+乙腈=65+35;
检测波长:300nm;
柱温:25℃;
进样量:10μL;
样品处理:50mg(精确到0.01mg)于10mL量瓶中,加入硫酸甲醇溶液溶解并定容至10mL。
HPLC谱图如图4所示。
实施例5
将实施例4中的滤液投入500ml四口瓶,于45℃,-0.1MPa条件下浓缩至120g左右,加入100g樟脑磺酸,搅拌至基本溶清。搅拌下往四口瓶加入65g30%氢氧化钠,调节PH=7-8。此时体系有少许不溶物,加入2g活性炭,室温搅拌30min后抽滤,澄清的淡黄色滤液搅拌升温至45℃。
控温45±2℃,往四口瓶缓慢滴加45g苯甲醛,约5h滴毕。滴毕控温45±2℃保温搅拌2h。
搅拌升温至50℃,用59g 50%硫酸缓慢调节PH=1后搅拌降温至≤10℃并控温≤10℃保温搅拌1h。
抽滤,滤饼用适量水洗涤,得157g亮白色结晶性固体粉末湿品和淡黄色滤液。
将所得湿品于80-90℃减压干燥至恒重,得135g亮白色结晶性固体粉末干品,每分子该产品含一分子结晶水。
用国食药监许[2010]456号附件中所述的亚苄基樟脑磺酸检测方法检测实施例5所得产品,具体结果如下:
HPLC条件:
色谱柱:C18柱,250mm×4.6mm,5μm;
流动相:20mmol/L的醋酸铵水溶液(乙酸调PH=5.1)+乙腈=65+35;
检测波长:300nm;
柱温:25℃;
进样量:10μL;
样品处理:50mg(精确到0.01mg)于10mL量瓶中,加入硫酸甲醇溶液溶解并定容至10mL。
HPLC谱图如图5所示。
本发明的反应方程式是:
本发明和现有技术相比,所涉及的亚苄基樟脑磺酸制备方法原料成本低,废水量少,安全性高,适用于工业化生产。
以上列举的仅是本发明的具体实施例子,本发明不限于以上实施例子,还可以有许多类似结构变化设计。本领域的普通技术人员能从本发明公开的内容直接导出或联想到的所有变形,均应认为是本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!