一种CUX1蛋白可结合DNA片段及在CUX1活性检测中的应用的制作方法
本发明涉及分子生物学领域,更特别地,涉及一种cux1蛋白可结合的dna片段及其在检测细胞内cux1转录调控活性中的应用。
背景技术:
cux1(ccaatdisplacementprotein),又名为cdp/cux/cut(ccaat-displacementprotein/cuthomeobox),是一类在多细胞物种中分布极其广泛的转录家族蛋白,在基因转录调控中发挥至关重要的作用,此外,该分子也参与基因转录后水平的调控,具有多种生物学功能。多项研究表明:cux1作为一个转录因子,可与多种原癌基因启动子区域(转录起始位点上游-2000bp左右)结合并调控其表达。
研究认为:cux1蛋白可通过其特有的dna结合域与多种蛋白质和dna结合,发挥其生物学功能。并且cux1的功能具有双面性,即可以激活也可以抑制基因启动子及转录,影响细胞的生长、增殖、分化和凋亡等过程。研究还发现,在表达过程中,cux1蛋白有不同的剪切变体,它们对靶基因的调控作用并不一致。cux1蛋白全长分子,即p200,与dna的结合具有不稳定性,多表现为转录抑制;而经过溶酶体组织蛋白酶l水解之后的截短产物,即p110等,其与靶基因的结合更稳定,多参与转录激活的过程,更多被当作一个转录因子进行研究。
cux1蛋白结合位点的改变可能造成其活性的改变,引起细胞功能紊乱,最终导致疾病的发生,甚至诱发恶性肿瘤。因此,检测cux1的转录调控活性对于探知细胞内部的病理发生机制十分重要。然而,目前检测cux1内源性活性仍然依靠westernblot或qrcr等方法,虽然灵敏度高,但检测到的只是cux1蛋白和mrna含量的差异,并不能直接体现其作为转录因子结合至靶序列以调控转录的活性改变。我们需要检测的不仅是ctcf的转录本或蛋白质含量,而是需要检测ctcf结合至有效位点以影响转录的活性。
因此,需要设计一种新的用于检测细胞内cux1转录调控活性的方法。
技术实现要素:
发明人通过研究发现,cux1蛋白结合的dna序列存在高度的保守性,其结合的dna核心序列为atcrat(r表示a、g)。
基于以上研究,本发明提供了一种cux1蛋白可结合的dna片段,其包含一个或多个cux1蛋白结合框,每个所述cux1蛋白结合框的序列独立地为5’-atcgat-3’和/或5’-atcaat-3’。
优选地,当包含多个cux1蛋白结合框时,每两个相邻的cux1蛋白结合框之间具有间隔序列,并且每两个相邻的cux1蛋白结合框之间的间隔序列各自独立地为3-10bp长。
优选地,序列如seqidno:1所示。
本发明还提供了上述dna片段在检测细胞内cux1转录调控活性中的应用。
本发明还提供了一种用于检测细胞内cux1转录调控活性的方法,其包括以下步骤:
s1:将包括上述dna片段以及连接于所述dna片段下游的报告基因表达框的报告基因系统导入所述细胞;
s2:通过检测所述报告基因的表达来计算所述细胞内cux1转录调控活性。
优选地,所述报告基因系统为双荧光素酶报告基因系统,包括重组质粒和对照质粒,并且所述重组质粒带有所述dna片段以及连接在所述dna片段下游的荧光素酶i的表达框,所述对照质粒带有荧光素酶ii的表达框,所示荧光素酶i与所述荧光素酶ii不同。
优选地,所述重组质粒通过将所述dna片段插入至质粒pgl3-basic的kpni与hindiii位点之间得到。
优选地,所述对照质粒为phrl-tk。
优选地,s1具体包括:
s11:将所述细胞培养至贴壁,并恢复形态;
s12:将所述重组质粒和对照质粒转染至细胞中。
优选地,s2具体包括:
s21:转染后将细胞继续培养24-36小时,洗涤细胞;
s22:分别检测转染后的细胞中的荧光素酶i和荧光素酶ii的活性;
s23:将荧光素酶ii活性归一化荧光素酶i活性得到值作为衡量cux1转录调控活性的指标。
通过使用本发明的dna片段和方法,可直接针对性地检测细胞内cux1的转录调控活性,而非仅仅检测其转录本或蛋白质的含量,从而使得能够更准确地分析cux1作为转录因子在一些病理学发展中所起的作用。
附图说明
图1为kpni与hindiii对重组质粒进行双酶切后的琼脂糖凝胶电泳照片;
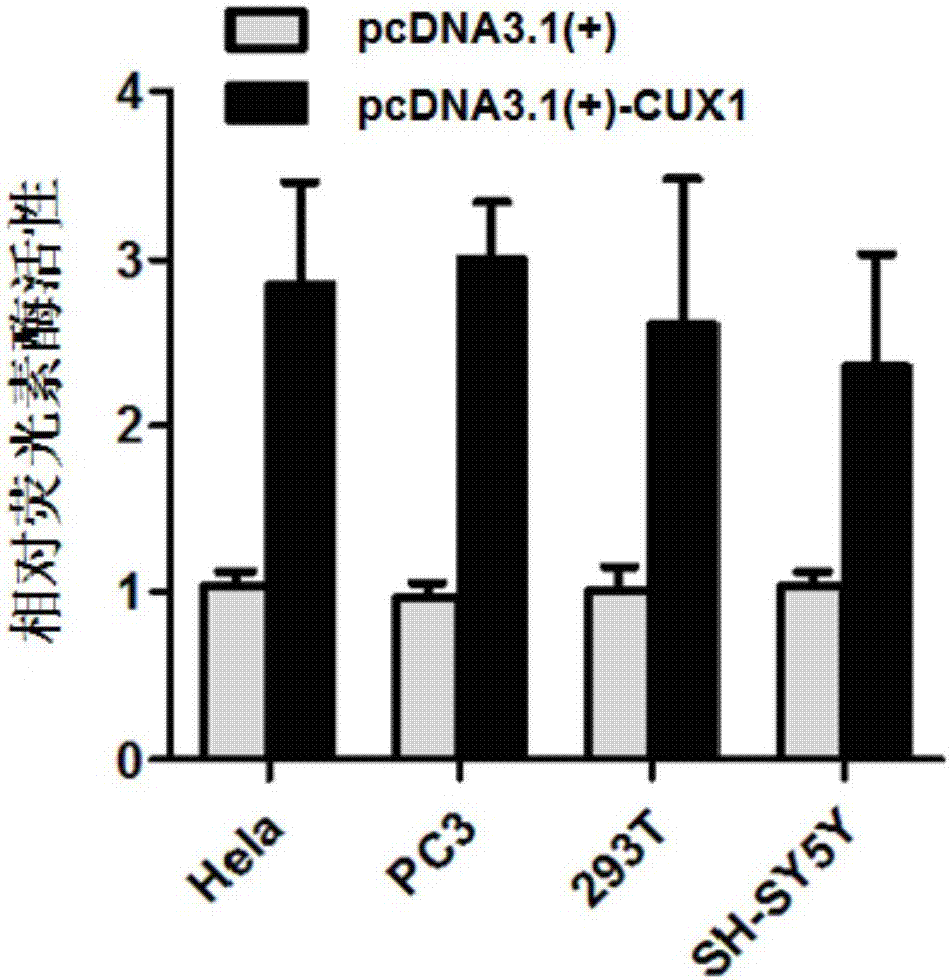
图2为转染有pgl3-basic-cux1和phrl-tk的人宫颈癌细胞系hela、人前列腺癌细胞系pc-3、人肾上皮细胞293t、人神经母细胞瘤细胞系sh-sy5y细胞中的萤火虫荧光素酶相对活性统计图,该相对活性通过将海肾荧光素酶活性归一化来得到。
具体实施方式
以下结合实例对本发明的原理和特征进行描述,所举实例只用于解释本发明,并非用于限定本发明的范围。
1.构建cux1蛋白可结合的dna片段
发明人对cux1进行研究分析,发现cux1蛋白结合的dna序列为atcrat(r表示a、g),合成该dna核心序列时,将这段dna核心序列重复四次,以核心碱基邻近碱基进行间隔,并连接至携带萤火虫荧光素酶的表达载体(pgl3.0-basic)中,构建成功后,该荧光素酶报告基因表达载体即包含四个重复的cux1蛋白结合的dna核心序列,以核心碱基临近的碱基进行间隔,其序列如seqidno:1所示。
为保证载体构建的成功,在末端加入相应的限制性内切酶酶切位点的粘性末端,本例在5’端加入kpni酶切位点的粘性末端(catgg),在3’端加入hindiii酶切位点的粘性末端(ttcga),序列两边的粘性末端的设计有助于片段与载体的连接。
通过从头合成的方法,分别合成该片段的两条寡核苷酸链,其序列分别如seqidno:2和3所示,这两条寡核苷酸链皆由武汉擎科生物技术有限公司合成。两条寡核苷酸链互补配对后形成包含kpni酶切位点粘性末端和hindiii酶切位点粘性末端的双链dna片段。100μl退火体系如下:annealingbufferfordnaoligos(5x)20μl、寡核苷酸链(50μm)各20μl,余下为ddh2o。
充分混匀后,设置pcr仪程序进行退火反应,具体程序为:95℃退火2分钟(目的是让dnaoligo充分变性);每90秒下降1℃,降至25℃后结束反应。dna退火产物用1.5%普通琼脂糖凝胶电泳鉴定,然后置冰上备用。
2.将cux1蛋白可结合的dna片段插入荧光素酶表达载体
用内切酶kpni和hindiii(购自takara公司,kpni货号为1618,hindiii货号为1615)对上述pgl3-basic空载体进行双酶切,20μl酶切体系如下:10xquickcutgreenbuffer2μl,kpni1μl,hindiii1μl,pgl3-basic空载体1μg,余下为ddh2o。
充分混匀后,37℃酶切反应90分钟,用1.5%普通琼脂糖凝胶电泳鉴定质粒双酶切后的情况,然后切胶回收(dna胶回收试剂盒购自天根生化科技有限公司,货号为dp209),回收结束后测样品浓度,置冰上备用。
将退火得到的双链dna连接至pgl3-basic荧光素酶表达载体上。10μl连接体系如下:dna连接酶(购自takara公司,货号为6022)5μl,双链dna片段与经双酶切的pgl3-basic载体共5μl。为促进酶连成功率,将dna片段与载体的摩尔比控制在10:1左右。充分混匀后,将酶连体系置于pcr仪中,16℃反应1小时,连接反应结束后得到重组质粒,置于冰上备用。
将重组质粒(含有cux1蛋白结合的dna片段的pgl3-basic表达载体)转化至大肠杆菌。按照经典的热激法进行转化,具体方法如下:从-80℃冰箱中取出大肠杆菌感受态细胞dh5α置于冰上解冻2-3分钟,在感受态处于将融未融的状态时,立即加入上述连接好的重组质粒,轻轻混匀,冰上孵育10-30分钟,然后42℃热激60秒,在立即放至冰上静置3-5分钟,最后加入500μl不含抗生素的lb液体培养基(经过高压灭菌处理),放置于恒温摇床中扩增1小时,扩增条件为37℃,200转/分钟。取扩增后的菌液100μl均匀涂布于直径为3.5厘米含氨苄青霉素的lb固体选择培养基中,室温静置10分钟后,倒置于37℃恒温培养箱中过夜。次日挑取单菌落至5ml含氨苄青霉素的lb液体培养基中,经37℃200转/分钟,摇菌繁殖12小时后提取质粒,1.5%普通琼脂糖凝胶电泳鉴定。
用内切酶kpni和hindiii对上述提取的质粒进行双酶切鉴定,本发明实施例双酶切鉴定如图1所示(泳道1为marker,泳道2-4均显示为阳性克隆)。阳性组送至武汉擎科生物技术有限公司测序,确认dna片段已整合至pgl3-basic载体上.至此,含有cux1蛋白结合的dna片段的pgl3-basic表达载体(以下均表示为pgl3-basic-cux1)构建成功。
3.pgl3-basic-cux1转染细胞
本发明实施例采用人宫颈癌细胞系hela、人前列腺癌细胞系pc3、人肾上皮细胞293t、人神经母细胞瘤细胞系sh-sy5y细胞进行双荧光素酶实验。分别将对数期生长的细胞hela、pc3、293t和sh-sy5y均匀种植在24孔板内,每孔约10万个细胞,用10%胎牛血清,高糖dmem培养基培养过夜。待细胞贴壁并恢复形态后,将报告基因系统相关质粒转染至细胞中(转染时细胞密度约为60-80%为宜,转染前2小时更换细胞培养基)。使用的转染试剂为neofect(购自零客创智生物科技有限公司,货号为tf20121201)。50μl转染体系如下:质粒0.5μg,neofect转染试剂0.5μl,余下为无血清培养基。
本实验中使用的质粒分别为:pgl3-basic空载体、pgl3-basic-cux1(含有cux1蛋白结合的dna核心序列的pgl3-basic表达载体)、phrl-tk(带有海肾荧光素酶活性作为内参素酶基因的质粒)、pcdna3.1(+)-cux1(cux1过表达质粒)。
4.计算cux1的活性
以海肾荧光素酶活性作为内参进行标准化,计算cux1活性。具体实验方法如下:转染后的细胞继续培养24-36小时,再弃培养基上清,用1xpbs清洗细胞3次,每次5分钟,清洗细胞时注意操作轻柔,避免正面吹打细胞。采用双荧光素酶报告基因检测试剂盒(购自盖宁生物科技有新公司,货号为gn201-01)进行检测。测定结果如图2所示,在人宫颈癌细胞系hela、人前列腺癌细胞系pc-3、人肾上皮细胞293t、人神经母细胞瘤细胞系sh-sy5y细胞中,cux1均具有较高的活性。
以上所述仅为本发明的较佳实施例,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
序列表
<110>华中科技大学同济医学院附属协和医院
<120>一种cux1蛋白可结合dna片段及在cux1活性检测中的应用
<130>1
<160>3
<170>patentinversion3.5
<210>1
<211>47
<212>dna
<213>人工序列
<400>1
tatatcgattatctcatcgatcatcttatcaattatctcatcgatta47
<210>2
<211>50
<212>dna
<213>人工序列
<400>2
ctatatcgattatctcatcgatcatcttatcaattatctcatcgattata50
<210>3
<211>58
<212>dna
<213>人工序列
<400>3
agcttataatcgatgagataattgataagatgatcgatgagataatcgatataggtac58
- 还没有人留言评论。精彩留言会获得点赞!