在pH响应再生型两水相体系中制备头孢丙烯的方法与流程
本发明涉及头孢丙烯技术领域,特别涉及头孢丙烯的合成方法技术领域,具体是一种在ph响应再生型两水相体系中制备头孢丙烯的方法。
背景技术:
头孢丙烯含有β-内酰胺环,属于β-内酰胺类抗生素,能抑制细菌细胞壁的合成。作为二代头孢菌素类抗生素,相比于一代抗生素而言,毒性低、活性强、抗菌谱广(很大程度的扩大了g-的抗菌范围,主要针对金黄色葡萄球菌和链球菌)等优点。
传统的化学合成头孢丙烯法存在工艺复杂、生产周期长、环境污染等不足,使得生产成本居高不下。水相酶促合成方法是实现头孢菌素反应温和、少步骤、更环保的生产途径之一,现行的酶法合成仍存在反应物价格高、转化率低、酶活性低、稳定性差等问题。
两水相体系避免了化学合成方法的高成本且可替代水相酶反应,将反应物与产物有效的分开,达到一边合成一边分离产物的效果,实现较高的产物转化率并在一定程度上分离纯化产物,且再生型两水相体系可以实现聚合物的高效回收。
技术实现要素:
本发明为了解决上述问题,提供一种能避免化学合成方法的高成本、改善单水相酶催化反应的低产物转化率、有效提高产率、简化操作、降低成本、轻易回收两水相体系的在ph响应再生型两水相体系中制备头孢丙烯的方法。
为了实现上述目的,本发明提供了一种在ph响应再生型两水相体系中制备头孢丙烯的方法,其主要特点是:
所述的方法包括以下步骤:
步骤(1):配制聚合物padba、padb、pmdb溶液,混合并构建两种两水相体系;
步骤(2):按照7-apra:d-对羟基苯甘氨酸甲酯盐酸盐摩尔比为1:1~1:4,固液比1:10~1:40依次加入7-apra、d-对羟基苯甘氨酸甲酯盐酸盐,调节溶液ph值至5.00~6.50、控制溶液温度为10℃~30℃;
步骤(3):将固定化青霉素酰化酶加入步骤(2)中获得的溶液中,搅拌反应2~20小时;
步骤(4):静置去除固定化青霉素酰化酶,调节反应液ph,回收两水相体系padba/pmdb、padb/pmdb聚合物,离心得上清液用于结晶,加入体积比为1:3~1:10的n,n-二甲基甲酰胺,调节结晶ph为5.00~8.00、温度0~10℃、结晶0.5~2.5小时,然后离心、洗涤、干燥得到产物。
较佳地,其特征在于,所述的步骤(1)中还包括加入盐溶液调节头孢丙烯的分配能力。
更佳地,所述的盐溶液为氯化钾或硫氰化钾溶液。
更佳地,所述的步骤(1)中头孢丙烯在两水相体系中的分配系数介于1.30~2.50之间。
较佳地,所述的聚合物padba、padb、pmdb溶液的浓度为6%~8%。
较佳地,所述的步骤(2)中添加7-apra后,先滴加浓氨水溶液,待两水相体系反应液澄清后再缓慢加入d-对羟基苯甘氨酸甲酯盐酸盐。
较佳地,所述的步骤(2)中固液比为1:20~1:30,7-apra:d-对羟基苯甘氨酸甲酯盐酸盐摩尔比为1:1.5~1:3.0。
较佳地,所述的步骤(3)中所述的固定化青霉素酰化酶的酶活为92u/g,加入的固定化青霉素酰化酶的量为2~10u/ml。
较佳地,所述的步骤(4)中所述的两水相体系padba/pmdb通过调节溶液ph至3.56得到回收,所述的两水相体系padb/pmdb通过调节溶液ph至3.54得到回收。
本发明提供的在ph响应再生型两水相体系中制备头孢丙烯的方法的有益效果在于:采用在ph响应型两水相体系中利用固定化青霉素酰化酶进行酶反应合成头孢丙烯,首先避免了化学合成方法的高成本;其次改善单水相酶催化反应的低产物转化率问题,两水相体系能很好的将产物与反应物分离,在促进酶反应的同时分离产物,可有效提高产率、简化操作、降低成本,通过本发明提供的制备头孢丙烯的方法头孢丙烯摩尔产率达到99.39%,相比于同等条件下的水相酶反应,产物的摩尔产率提高了28.06%,产物纯度达到88.02%;此外,在反应结束后的聚合物可以通过简单的酸碱调节得到回收。
附图说明
图1为本发明实施例1、2的头孢丙烯动态光散射结果。
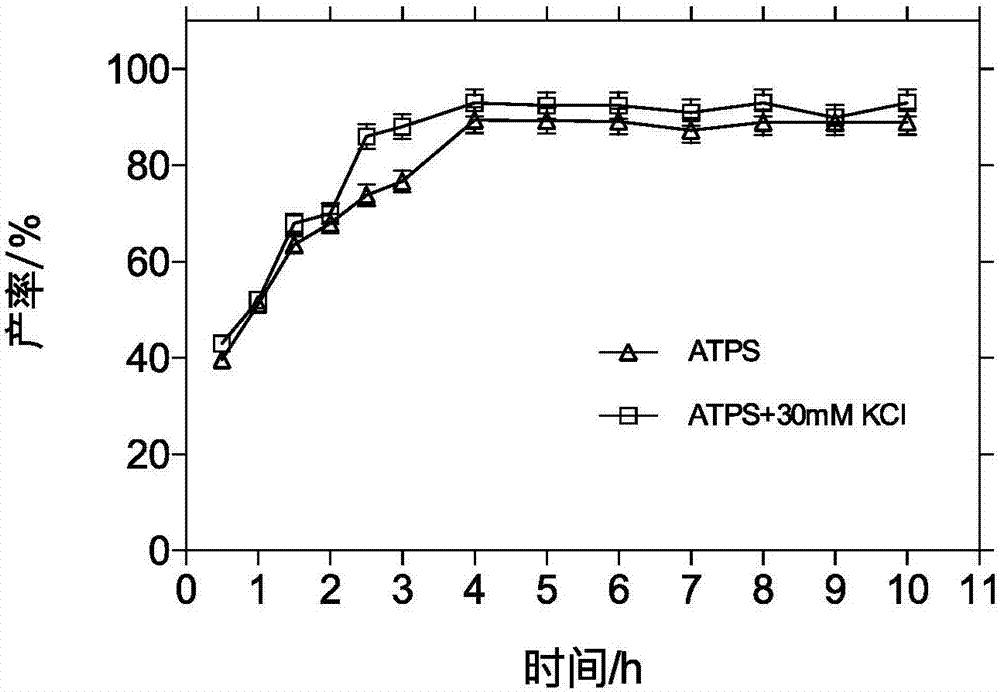
图2为本发明实施例1、2中的padba/pmdb两水相体系中酶反应相转移合成头孢丙烯摩尔产率随时间变化示意图。
图3为本发明实施例5、6中的padb/pmdb两水相体系中酶反应相转移合成头孢丙烯酶产率随时间变化示意图。
具体实施方式
下面结合附图对本发明的具体实施方法作进一步说明。
本发明提供的方法包括如下步骤:
(1)、配制浓度为6~8%(w/v)的聚合物padba、padb、pmdb溶液,混合构建两种两水相体系,加入盐离子调节物质在两相中的分配能力。
(2)、按照母核侧链摩尔比1:1~1:4,固液比1:10~1:40依次加入7-apra,d-对羟基苯甘氨酸甲酯盐酸盐,调节溶液ph为5.00-6.50、控制温度10~30℃、加入2~10u/ml固定化青霉素酰化酶、反应2~20小时。
(3)、静置去除固定化酶,调节反应液ph回收两水相padba/pmdb,padb/pmdb聚合物,离心得上清液用于结晶。加入体积比为1:3~1:10的n,n-二甲基甲酰胺,调节结晶溶液ph为5.00-8.00、温度0~10℃、结晶0.5~2.5小时,然后离心、洗涤、干燥得到产物。
其中在(1)中,酶反应在两水相中进行,本发明提供一种较优的两水相体系,分别为padba3%(w/v)/pmdb3%(w/v)、padb4%(w/v)/pmdb3%(w/v),其中padba的制备可参见中国专利cn102344520a,padb及pmdb的制备请参见中国专利cn102875721a。
其中在(2)中,7-apra为具有如下结构式(i)的化合物,即,7-氨基-3-丙烯基头孢烷酸:
所述d-对羟基苯甘氨酸甲酯盐酸盐(d-hpgme-hcl)为具有如下结构式(ii)的化合物:
本发明提供的在ph响应再生型两水相体系中制备头孢丙烯的反应式如下所示,其中反应时中的结构(iii)为最终得到的头孢丙烯:
此外,步骤(1)中,加入的盐离子为20~60mm的氯化钾或硫氰化钾,例如,浓度可以分别为30mm氯化钾(padba/pmdb)、40mm硫氰化钾(padb/pmdb)。
步骤(2)中,添加7-apra后,可以先滴加浓氨水促溶,待两水相体系反应液澄清后再缓慢加入d-hpgme-hcl,防止局部过酸析出聚合物和7-apra。
本发明提供一种方法的步骤(2)中的固液比和母核侧链的实施例,其中,固液比为1:20~1:30,母核侧链摩尔比为1:1.5~1:3.0。
步骤(2)中,酶反应的ph为5.00~6.00、控制温度15~25℃,例如温度可以为20℃。
步骤(2)中,加入固定化青霉素酰化酶4.6u/ml,反应3~10小时。
步骤(3)中所述的两水相体系padba/pmdb、padb/pmdb可通过调节溶液ph得到回收。
步骤(3)中,两水相体系padba/pmdb在ph为3.56时回收率达到94.33%,padb/pmdb体系ph在3.54,回收率达到96.01%。
步骤(3)中,n,n-二甲基甲酰胺添加体积比为1:5~1:7。
步骤(3)中,调节结晶ph为6.00~7.00、温度0~4℃、结晶1~2小时,离心、洗涤、干燥得到产物。
一般用分配系数(k)来衡量一种物质在某一体系中的分配能力,其中分配系数(k)通常利用下述公式进行计算:
k=ctop/cbottom,
其中,式中的k为被分配物质的分配系数,
ctop表示的为被分配物质在上相中的浓度;
cbottom表示的为被分配物质在下相中的浓度。
实施例1
padba/pmdb两水相体系中酶反应相转移合成头孢丙烯:
配置padba3%(w/v)/pmdb3%(w/v)两水相体系20ml,将0.2700g7-氨基-3-丙烯基头孢烷酸(7-apra)和0.7320gd-对羟基苯甘氨酸甲酯盐酸盐(d-hpgme-hcl)加入两水相体系中,加入0.0136g磷酸二氢钾,调节溶液ph至5.50,控制反应温度为20℃,加入1g固定化青霉素酰化酶(ipga),控制反应转速200r/min,反应4小时,通过高效液相色谱法检测得到头孢丙烯摩尔产率为89.41%,头孢丙烯分配系数k保持在1.30,调节溶液ph回收两水相聚合物,回收率达到94.33%,加入5倍体积dmf结晶、离心、洗涤、干燥得到粗产品头孢丙烯。
经检测得到如图1所示的产物的动态光散射图,其中横坐标material分别为头孢丙烯不同条件下得到的产品,纵坐标radiums为粒子半径(nm):
由图1可知:a,b,c分别代表了头孢丙烯标准品,水相结晶头孢丙烯,两水相结晶头孢丙烯。与a组相比,*p<0.05,**p<0.01。物质粒径结果表现为平均值±sd。对于产物头孢丙烯与纯品粒径的比较发现,纯品头孢丙烯的粒径在0.568nm,水相结晶的头孢丙烯粒径在0.635nm,两水相结晶的头孢丙烯粒径在0.577nm,差异不显著。可以证明,两水相中的聚合物在酶反应结束后被全部的去除干净,上清液用于头孢丙烯的结晶,结晶产物中没有聚合物的残留,同时可以实现聚合物的回收再利用。
实施例2
padba/pmdb两水相体系中酶反应相转移合成头孢丙烯:
配置padba3%(w/v)/pmdb3%(w/v)两水相体系20ml,将0.2700g7-apra和0.7320gd-hpgme-hcl加入两水相体系中,加入0.0136g磷酸二氢钾及0.0447g氯化钾,调节溶液ph至5.50,控制反应温度为20℃,加入1gipga,控制反应转速200r/min,反应4小时,头孢丙烯分配系数k保持在2.13,通过液相检测得到头孢丙烯摩尔产率为93.02%,提高了16.48%。调节溶液ph回收两水相聚合物,回收率达到94.33%,加入5倍体积dmf结晶、离心、洗涤、干燥得到粗产品头孢丙烯。
经检测得到的头孢丙烯粒子半径与实例1相同。
如图2所示为实施例1~2的padba/pmdb两水相体系中酶反应相转移合成头孢丙烯摩尔产率随时间变化示意图。在反应4小时头孢丙烯摩尔产率达到89.41%,加入30mmkcl后,产率为93.02%,水相酶反应摩尔产率最高为79.85%。对于水相反应,在达到最高产率后反应随着时间的进行产物开始部分水解,两水相能解决产物水解作用且加入一定浓度的盐离子可提高产物产率。
实施例3
padb/pmdb两水相体系中酶反应相转移合成头孢丙烯:
配置padb4%(w/v)/pmdb3%(w/v)两水相体系20ml,将0.2831g7-apra和0.3835gd-hpgme-hcl加入两水相体系中,加入0.0136g磷酸二氢钾,调节溶液ph至5.40,控制反应温度为20℃,加入ipga1g,控制反应转速200r/min,反应4小时,通过液相检测得到头孢丙烯摩尔产率为80.19%,头孢丙烯分配系数k保持在1.30。调节溶液ph回收两水相聚合物,回收率达到96.01%,加入5倍体积dmf结晶、离心、洗涤、干燥得到粗产品头孢丙烯。
经检测得到的头孢丙烯粒子半径与实例1相同。
实施例4
padb/pmdb两水相体系中酶反应相转移合成头孢丙烯:
配置padb4%(w/v)/pmdb3%(w/v)两水相体系20ml,将0.2046g7-apra和0.4620gd-hpgme-hcl加入两水相体系中,加入0.0136g磷酸二氢钾,调节溶液ph至5.40,控制反应温度为20℃,加入1gipga,控制反应转速200r/min,反应7小时,通过液相检测得到头孢丙烯摩尔产率为98.80%,头孢丙烯分配系数k保持在1.30。调节溶液ph回收两水相聚合物,回收率达到96.01%,加入5倍体积dmf结晶、离心、洗涤、干燥得到粗产品头孢丙烯。
经检测得到的头孢丙烯粒子半径与实例1相同。
实施例5
padb/pmdb两水相体系中酶反应相转移合成头孢丙烯:
配置padb4%(w/v)/pmdb3%(w/v)两水相体系20ml,将0.2370g7-apra和0.4290gd-hpgme-hcl加入两水相体系中,加入0.0136g磷酸二氢钾,调节溶液ph至5.40,控制反应温度为20℃,加入1gipga,控制反应转速200r/min,反应7小时,头孢丙烯分配系数k保持在1.84。通过液相检测得到头孢丙烯摩尔产率为97.38%,提高了25.47%。调节溶液ph回收两水相聚合物,回收率达到96.01%,加入5倍体积dmf结晶、离心、洗涤、干燥得到粗产品头孢丙烯。
经检测得到的头孢丙烯粒子半径与实例1相同。
实施例6
padb/pmdb两水相体系中酶反应相转移合成头孢丙烯:
配置padb4%(w/v)/pmdb3%(w/v)两水相体系20ml,将0.2370g7-apra和0.4290gd-hpgme-hcl加入两水相体系中,加入0.0136g磷酸二氢钾及0.0776g硫氰化钾,调节溶液ph至5.40,控制反应温度为20℃,加入1gipga,控制反应转速200r/min,反应6小时,头孢丙烯分配系数k保持在1.84。通过液相检测得到头孢丙烯摩尔产率为99.39%,提高了28.06%。调节溶液ph回收两水相聚合物,回收率达到96.01%,加入5倍体积dmf结晶、离心、洗涤、干燥得到粗产品头孢丙烯。
经检测得到的头孢丙烯粒子半径与实例1相同。
如图3所示为实施例5~6的padb/pmdb两水相体系中酶反应相转移合成头孢丙烯酶产率随时间变化示意图。头孢丙烯的摩尔产率在7h达到最高,为97.38%,在加入40mmkscn后反应在6h达到99.39%,相同条件下水相酶反应产物产率在3h达到,为77.61%。两水相的优势在于,酶反应产率呈现持续上升趋势,且到达反应平衡后产物水解作用不明显,产率远高于水相酶反应。
综上,ph响应型可回收两水相体系可以达到分离纯化产物的作用,还可以解除酶反应的产物抑制,对提高产物头孢丙烯的产率有明显的效果且聚合物回收后对产物纯度没有影响。
本发明提供的在ph响应再生型两水相体系中制备头孢丙烯的方法,采用在ph响应型两水相体系中利用固定化青霉素酰化酶进行酶反应合成头孢丙烯,首先避免了化学合成方法的高成本;其次改善单水相酶催化反应的低产物转化率问题,两水相体系能很好的将产物与反应物分离,在促进酶反应的同时分离产物,可有效提高产率、简化操作、降低成本,通过本发明提供的制备头孢丙烯的方法头孢丙烯摩尔产率达到99.39%,与相同条件下水相酶反应产率相比提高了28.06%,产物纯度达到88.02%;此外,在反应结束后的聚合物可以通过简单的酸碱调节得到回收。
在此说明书中,本发明已参照其特定的实施例作了描述。但是,很显然仍可以做出各种修改和变换而不背离本发明的精神和范围。因此,说明书和附图应被认为是说明性的而非限制性的。
- 还没有人留言评论。精彩留言会获得点赞!