4‑((4‑取代芳基‑2‑嘧啶基)氨基)苯甲酰肼衍生物及其制备方法和应用与流程
本发明涉及肿瘤药物,尤其是涉及4-((4-取代芳基-2-嘧啶基)氨基)苯甲酰肼衍生物及其制备方法和应用。
背景技术:
肿瘤药物治疗开始于20世纪40年代,经过70年发展,目前全球临床应用的抗肿瘤药物约100种。进入21世纪,随着现代医学的发展、肿瘤分子机制研究的深入、现代生物医药技术的成熟,全球抗肿瘤药物研发硕果累累。据不完全统计,目前全球正处于临床研究阶段的抗肿瘤候选新药有450多种,共涉及2850余项临床研究,其中ⅲ期临床研究共223项,涉及新药80余种。抗肿瘤药物种类繁多,但是理想的肿瘤药物较少,现存的抗癌药物或多或少都有着生物利用度低、毒副作用大等缺点。因此设计合成具有生物利用度高、抗癌活性明显和毒性低等优点的抗肿瘤药物依旧是目前研究的热点。
技术实现要素:
本发明的第一目的在于提供4-((4-取代芳基-2-嘧啶基)氨基)苯甲酰肼衍生物。
本发明的第二目的在于提供4-((4-取代芳基-2-嘧啶基)氨基)苯甲酰肼衍生物的制备方法。
本发明的第三目的在于提供一类4-((4-取代芳基-2-嘧啶基)氨基)苯甲酰肼衍生物用于制备治疗或预防肿瘤相关疾病的药物。
所述4-((4-取代芳基-2-嘧啶基)氨基)苯甲酰肼衍生物的结构式为:
其中,r1代表c1~c4的直链烷基,c5或c6的环烷基,未取代的芳香烷基,取代苯基;r2代表cl或者h;x代表o或s;y代表n或c。
所述4-((4-取代芳基-2-嘧啶基)氨基)苯甲酰肼衍生物的制备方法包括以下步骤:
1)制备中间体对胍基苯甲酸乙酯硝酸盐;
2)制备中间体1-(取代芳基)-3-(二甲氨基)-2-丙烯-1-酮;
3)制备中间体4-((4-取代芳基-2-嘧啶基)氨基)苯甲酸乙酯;
4)制备中间体4-((4-取代芳基-2-嘧啶基)氨基)苯甲酰肼;
5)制备4-((4-取代芳基-2-嘧啶基)氨基)苯甲酰肼衍生物。
在步骤1)中,所述制备中间体对胍基苯甲酸乙酯硝酸盐的具体方法可为:在干燥的反应瓶中依次加入对氨基苯甲酸乙酯、氰胺和无水乙醇;在搅拌状态下加入盐酸后,升温至回流,反应22~24h;薄层色谱(tlc)检测反应后,停止反应;反应液减压浓缩除溶剂后加入水,在0℃加硝酸铵的水溶液,加入后保温反应,过滤,收集滤饼,烘干得白色固体产物对胍基苯甲酸乙酯硝酸盐,收率75%~80%;所述对氨基苯甲酸乙酯、氰胺、盐酸、硝酸铵的摩尔比可为1︰(2.5~3)︰(1.2~1.5)︰(2~2.5)。
在步骤2)中,所述制备中间体1-(取代芳基)-3-(二甲氨基)-2-丙烯-1-酮的具体方法可为:在干燥的反应瓶里依次加入2-乙酰吡啶(或3-乙酰吡啶,4-乙酰吡啶,4-氯苯乙酮),n,n-二甲基甲酰胺二甲基缩醛,加热至110℃,回流反应18~20h,tlc检测反应后,停止反应,反应液减压浓缩后加乙酸乙酯,搅拌,抽滤,收集滤饼,烘干得固体产物1-(取代芳基)-3-(二甲氨基)-2-丙烯-1-酮,收率56%~60%;所述2-乙酰吡啶(或3-乙酰吡啶,4-乙酰吡啶,4-氯苯乙酮)、n,n-二甲基甲酰胺二甲基缩醛的摩尔比可为1︰(1.1~1.2)。
在步骤3)中,所述制备中间体4-((4-取代芳基-2-嘧啶基)氨基)苯甲酸乙酯的具体方法可为:在干燥的反应瓶里依次加入对胍基苯甲酸乙酯硝酸盐、1-(取代芳基)-3-(二甲氨基)-2-丙烯-1-酮、氢氧化钠固体、无水乙醇,加热至回流,反应18~22h,tlc检测反应后,停止反应,冷却至室温后抽滤,收集滤饼,烘干,得固体产物4-((4-取代芳基-2-嘧啶基)氨基)苯甲酸乙酯,收率84%~90%;所述述对胍基苯甲酸乙酯硝酸盐、1-(取代芳基)-3-(二甲氨基)-2-丙烯-1-酮、氢氧化钠的摩尔比可为1︰(1~1.1)︰(1.2~1.5)。
在步骤4)中,所述制备中间体4-((4-取代芳基-2-嘧啶基)氨基)苯甲酰肼的具体方法可为:在干燥的反应瓶中依次加入4-((4-取代芳基-2-嘧啶基)氨基)苯甲酸乙酯、水合肼、无水乙醇,加热至回流,反应18~20h,tlc检测反应后,停止反应,冷却至室温后抽滤,收集滤饼,烘干,得固体产物4-((4-取代芳基-2-嘧啶基)氨基)苯甲酰肼,收率66%~70%;所述4-((4-取代芳基-2-嘧啶基)氨基)苯甲酸乙酯与水合肼的摩尔比可为1︰(18~20)。
在步骤5)中,所述制备4-((4-取代芳基-2-嘧啶基)氨基)苯甲酰肼衍生物的具体方法可为:在干燥的反应瓶里依次加入4-((4-取代芳基-2-嘧啶基)氨基)苯甲酰肼固体、n,n-二甲基甲酰胺,加热至100℃,待固体溶解完全,降温至70℃,加入异氰酸酯(可为脂肪异氰酸酯,取代或未取代的苯基异氰酸酯,取代或未取代的苯基硫代异氰酸酯,未取代的芳香烷基异氰酸酯,未取代的芳香烷基硫代异氰酸酯),保温反应3~4h;tlc检测反应后,停止反应,冷却至室温后加水,抽滤,收集滤饼,烘干,得到的粗品经硅胶柱层析分离(洗脱剂按体积比为二氯甲烷︰甲醇=50︰1)得到固体产物4-((4-取代芳基-2-嘧啶基)氨基)苯甲酰肼衍生物,收率56%~60%;所述4-((4-取代芳基-2-嘧啶基)氨基)苯甲酰肼、异氰酸酯的摩尔比可为1︰(1~1.1)。
所述一类4-((4-取代芳基-2-嘧啶基)氨基)苯甲酰肼衍生物用于制备治疗或预防肿瘤相关疾病的药物中的用途。
所述一类4-((4-取代芳基-2-嘧啶基)氨基)苯甲酰肼衍生物具有显著的抑制肿瘤细胞生长和诱导肿瘤细胞凋亡的作用,可用于制备治疗或预防肿瘤相关疾病的药物。
本发明的一类4-((4-取代芳基-2-嘧啶基)氨基)苯甲酰肼衍生物,其合成反应成本低,反应过程简单易控制,适用于工业化生产,且该类衍生物具有显著的抑制肿瘤细胞生长和诱导肿瘤细胞凋亡的作用,可在制备抗肿瘤药物上广泛应用。
附图说明
图1为n-环己基-2-(4–((4-(4-吡啶基)-2-嘧啶基)氨基)苯甲酰基)肼-1-甲酰胺对肿瘤细胞hepg2的生长抑制效果图。在图1中,横坐标为化合物作用时间(h),纵坐标为生长抑制率(%),设置浓度梯度为:曲线a为5μm/l,b为10μm/l,c为20μm/l,作用时间点分别为24h,48h,72h和96h。
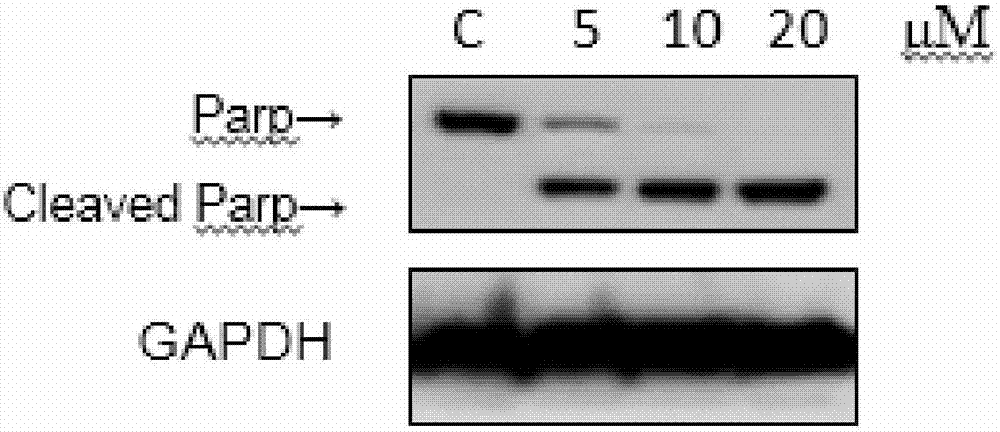
图2为n-环己基-2-(4–((4-(4-吡啶基)-2-嘧啶基)氨基)苯甲酰基)肼-1-甲酰胺引起hepg2细胞parp切割的wb检测图。在图2中,化合物的浓度梯度为5μm/l,10μm/l和20μm/l,对照组加入相应的dmso做对照,gapdh为内参。
具体实施方式
为了便于理解本发明,现结合具体实施方式对本发明作进一步说明,以进一步诠释本发明,但不构成对本发明的任何方式的限制。
实施例1:对胍基苯甲酸乙酯硝酸盐的合成
在200ml干燥的单颈瓶中,首先依次加入对氨基苯甲酸乙酯(5.0g,0.030mol),氰胺(50%w/w,7.6g,0.091mol),无水乙醇(20ml);再在搅拌状态下滴加浓盐酸(3.8ml,0.045mol),滴加结束后升温至回流,反应24h后,tlc检测反应已完全,停止反应。反应液减压浓缩除溶剂后加入50ml水,再在0℃下滴加硝酸铵(4.8g,0.061mol)的水溶液,滴加结束后保温反应1h,过滤,收集滤饼,烘干得白色固体产物对胍基苯甲酸乙酯硝酸盐6.3g,收率78%。
实施例2:1-(2-吡啶基)-3-(二甲氨基)-2-丙烯-1-酮的合成
在100ml干燥的单颈瓶里依次加入2-乙酰吡啶(5.0g,0.041mol),n,n-二甲基甲酰胺二甲基缩醛(5.4g,0.045mol),加热至110℃,回流反应18h后,tlc检测反应已完全,停止反应。反应液减压浓缩后加5ml乙酸乙酯,室温搅拌1h,抽滤,收集滤饼,烘干得黄绿色固体4.2g,收率58%。波谱数据:1hnmr(600mhz,dmso-d6):8.55~8.69(m,1h),7.98(d,j=7.7hz,1h),7.91(dt,j=1.7,7.66hz,1h),7.80(d,j=12.6hz,1h),7.50(ddd,j=1.2,4.77,7.52hz,1h),6.38(d,j=10.6hz,1h),3.18(s,3h),2.92(s,3h).
实施例3:4-((4-(2-吡啶基)-2-嘧啶基)氨基)苯甲酸乙酯的合成
在50ml干燥的单颈瓶中依次加入1-(2-吡啶基)-3-(二甲氨基)-2-丙烯-1-酮(1.00g,0.0056mol),对胍基苯甲酸乙酯硝酸盐(1.53g,0.0056mol),氢氧化钠固体(0.273g,0.0068mol),无水乙醇(10ml),加热至回流,反应18h后,tlc检测反应已完全,停止反应。冷却至室温后抽滤,收集滤饼,烘干,得灰白色固体1.52g,收率84%。波谱数据:1hnmr(600mhz,cdcl3):8.73(d,j=4.4hz,1h),8.62(d,j=4.9hz,1h),8.42(d,j=7.7hz,1h),8.07(d,j=8.2hz,2h),7.89-7.91(m,1h),7.87(d,j=5.6hz,1h),7.81(d,j=8.0hz,2h),7.56(br.s.,1h),7.40~7.45(m,1h),4.38(q,j=7.0hz,2h),1.41(t,j=6.9hz,3h).
实施例4:4-((4-(2-吡啶基)-2-嘧啶基)氨基)苯甲酰肼的合成
在50ml干燥的单颈瓶中依次加入4-((4-(2-吡啶基)-2-嘧啶基)氨基)苯甲酸乙酯(1.52g,0.0047mol),水合肼(5.00ml),无水乙醇(5ml),加热至回流,反应20h后,tlc检测反应已完全,停止反应。冷却至室温后抽滤,收集滤饼,烘干,得粉红色固体产物1.7g。收率70%。波谱数据:1hnmr(600mhz,dmso-d6):10.06(br.s.,1h),9.62(br.s.,1h),8.77(d,j=2.9hz,1h),8.70(d,j=4.7hz,1h),8.43(d,j=7.5hz,1h),8.07(t,j=7.4hz,1h),7.93(d,j=8.4hz,2h),7.84(d,j=8.2hz,2h),7.79(d,j=4.7hz,1h),7.54~7.64(m,1h),4.50(br.s.,2h).
实施例5:4-((4-取代芳基-2-嘧啶基)氨基)苯甲酰肼衍生物的制备:n-叔丁基-2-(4–((4-(2-吡啶基)-2-嘧啶基)氨基)苯甲酰基)肼-1-甲酰胺
在50ml干燥的双颈瓶里依次加入4-((4-(2-吡啶基)-2-嘧啶基)氨基)苯甲酰肼(0.100g,0.00032mol),n,n-二甲基甲酰胺(5ml),加热至100℃,待固体溶解完全,降温至70℃,加入叔丁基异氰酸酯(0.032g,0.00035mol),保温反应3h。tlc检测反应完全,停止反应。冷却至室温后加10ml水,抽滤,收集滤饼,烘干,得到的粗品经硅胶柱层析分离(洗脱剂为二氯甲烷:甲醇=50︰1,v/v)得到黄色固体产物0.090g,收率68%。波谱数据:1hnmr(600mhz,dmso-d6):10.10(s,1h),9.95(d,j=2.0hz,1h),8.76(d,j=4.0hz,1h),8.71(d,j=4.9hz,1h),8.43(d,j=7.8hz,1h),8.07(dt,j=1.6,7.70hz,1h),7.93~7.96(m,2h),7.87-7.90(m,2h),7.80(d,j=4.9hz,1h),7.58~7.61(m,2h),6.08(br.s.,1h),1.27(s,9h).13cnmr(151mhz,dmso-d6):166.3,163.4,160.2,160.1,157.8,153.9,150.2,144.1,138.1,128.7,126.3,125.6,121.6,118.2,109.2,49.9,29.6.hrms(+):calcdforc21h24n7o2+[m+h]+406.1986,found406.1982;calcdforc21h23n7o2na+[m+na]+428.1805,found428.1802.
实施例6:4-((4-取代芳基-2-嘧啶基)氨基)苯甲酰肼衍生物的制备:n-苯基-2-(4–((4-(2-吡啶基)-2-嘧啶基)氨基)苯甲酰基)肼-1-硫代甲酰胺
在50ml干燥的双颈瓶里依次加入4-((4-(2-吡啶基)-2-嘧啶基)氨基)苯甲酰肼(0.100g,0.00032mol),n,n-二甲基甲酰胺(5ml),加热至100℃,待固体溶解完全,降温至70℃,加入硫代异氰酸苯酯(0.044g,0.00035mol),保温反应3h。tlc检测反应已完全,停止反应。冷却至室温后加10ml水,抽滤,收集滤饼,烘干,得到的粗品经硅胶柱层析分离(洗脱剂为二氯甲烷:甲醇=50︰1,v/v)得到白色固体产物0.070g,收率50%。波谱数据:1hnmr(600mhz,dmso-d6):10.39(s,1h),10.13(s,1h),9.80(br.s.,1h),9.68(s,1h),8.76~8.78(m,1h),8.72(d,j=4.9hz,1h),8.43(d,j=7.8hz,1h),8.07(dt,j=1.7,7.7hz,1h),7.98(s,4h),7.81(d,j=5.1hz,1h),7.59(ddd,j=1.1,4.7,7.5hz,1h),7.46(d,j=1.8hz,2h),7.34(t,j=7.7hz,2h),7.13~7.19(m,1h).13cnmr(151mhz,dmso-d6):166.1,163.4,160.2,160.1,153.9,150.2,144.3,139.8,138.1,129.3,128.4,126.5,126.3,125.5,125.4,121.6,118.0,109.3.hrms(+):calcdforc23h20n7os+[m+h]+442.1445,found442.1444,calcdforc23h19n7osna+[m+na]+464.1264,found464.1265.
实施例7:4-((4-取代芳基-2-嘧啶基)氨基)苯甲酰肼衍生物的制备:n-(3-氯-4-甲基苯
基)-2-(4–((4-(4-吡啶基)-2-嘧啶基)氨基)苯甲酰基)肼-1-甲酰胺
在50ml干燥的双颈瓶里依次加入4-((4-(4-吡啶基)-2-嘧啶基)氨基)苯甲酰肼(0.150g,0.00048mol),n,n-二甲基甲酰胺(7ml),加热至100℃,待固体溶解完全,降温至70℃,加入3-氯-4-甲基苯基异氰酸酯(0.090g,0.00054mol),保温反应3h。tlc检测反应完全,停止反应。冷却至室温后加10ml水,抽滤,收集滤饼,烘干,得到的粗品经硅胶柱层析分离(洗脱剂为二氯甲烷:甲醇=50︰1,v/v)得到黄色固体产物0.120g,收率51%。波谱数据:1hnmr(600mhz,dmso-d6):10.18(s,1h),10.15(s,1h),8.97(br.s.,1h),8.81(br.s.,2h),8.73(d,j=5.1hz,1h),8.23(br.s.,1h),8.12(d,j=5.8hz,2h),7.95~7.98(m,2h),7.91~7.94(m,2h),7.68(d,j=1.6hz,1h),7.62(d,j=4.9hz,1h),7.29(br.s.,1h),7.22(d,j=8.2hz,1h),2.25(s,3h).13cnmr(151mhz,dmso-d6):166.7,162.0,160.5,160.4,151.0,144.3,144.1,139.4,133.4,131.5,128.9,128.7,125.6,121.5,118.3,109.8,19.2.hrms(+):calcdforc24h21cln7o2+[m+h]+474.1440,found474.1440;calcdforc24h20cln7o2na+[m+na]+496.1259,found496.1259.
实施例8:4-((4-取代芳基-2-嘧啶基)氨基)苯甲酰肼衍生物的制备:n-环己基-2-(4–((4-(4-吡啶基)-2-嘧啶基)氨基)苯甲酰基)肼-1-甲酰胺
在50ml干燥的双颈瓶里依次加入4-((4-(4-吡啶基)-2-嘧啶基)氨基)苯甲酰肼(0.100g,0.00033mol),n,n-二甲基甲酰胺(5ml),加热至100℃,待固体溶解完全,降温至70℃,加入3-氯-4-甲基苯基异氰酸酯(0.045g,0.00036mol),保温反应3h。tlc检测反应已完全,停止反应。冷却至室温后加10ml水,抽滤,收集滤饼,烘干,得到的粗品经硅胶柱层析分离(洗脱剂为二氯甲烷:甲醇=50︰1,v/v)得到白色固体产物0.070g,收率50%。波谱数据:10.18(s,1h),10.15(s,1h),8.97(br.s.,1h),8.81(br.s.,2h),8.73(d,j=5.1hz,1h),8.23(br.s.,1h),8.12(d,j=5.8hz,2h),7.95~7.98(m,2h),7.91~7.94(m,2h),7.68(d,j=1.6hz,1h),7.62(d,j=4.9hz,1h),7.29(br.s.,1h),7.22(d,j=8.2hz,1h),2.25(s,3h).13cnmr(151mhz,dmso-d6):166.4,162.0,160.4,158.2,151.1,144.2,143.9,128.8,125.7,121.4,118.3,109.8,48.6,33.5,25.7,25.1.hrms(+):calcdforc23h26n7o2+[m+h]+432.2142,found432.2141;calcdforc23h25n7o2na+[m+na]+454.1962,found454.1960.
实施例9:4-((4-取代芳基-2-嘧啶基)氨基)苯甲酰肼衍生物的制备:n-丁基-2-(4–((4-(4-吡啶基)-2-嘧啶基)氨基)苯甲酰基)肼-1-甲酰胺
在50ml干燥的双颈瓶里依次加入4-((4-(4-吡啶基)-2-嘧啶基)氨基)苯甲酰肼(0.100g,0.00033mol),n,n-二甲基甲酰胺(5ml),加热至100℃,待固体溶解完全,降温至70℃,加入丁基异氰酸酯(0.035g,0.00036mol),保温反应3h。tlc检测反应已完全,停止反应。冷却至室温后加10ml水,抽滤,收集滤饼,烘干,得到的粗品经硅胶柱层析分离(洗脱剂为二氯甲烷:甲醇=50︰1,v/v)得到淡黄色固体产物0.080g,收率60%。波谱数据:1hnmr(600mhz,dmso-d6):10.16(s,1h),9.96(s,1h),8.80~8.83(m,2h),8.73(d,j=5.1hz,1h),8.12~8.14(m,2h),7.93~7.95(m,2h),7.89~7.92(m,2h),7.74(s,1h),7.62(d,j=5.1hz,1h),6.45(br.s.,1h),3.03(q,j=6.7hz,2h),1.38(quin,j=7.2hz,2h),1.28(sxt,j=7.3hz,2h),0.87(t,j=7.3hz,3h).13cnmr(151mhz,dmso-d6):166.5,161.9,160.5,159.0,150.9,144.4,143.9,128.9,125.8,121.5,118.3,109.8,39.3,32.5,19.9,14.2.hrms(+):calcdforc21h24n7o2+[m+h]+406.1986,found406.1985;calcdforc21h23n7o2na+[m+na]+428.1805,found428.1806.
实施例10:4-((4-取代芳基-2-嘧啶基)氨基)苯甲酰肼衍生物的制备:n-苯乙基-2-(4–((4-(3-吡啶基)-2-嘧啶基)氨基)苯甲酰基)肼-1-甲酰胺
在50ml干燥的双颈瓶里依次加入4-((4-(3-吡啶基)-2-嘧啶基)氨基)苯甲酰肼(0.150g,0.00049mol),n,n-二甲基甲酰胺(7ml),加热至100℃,待固体溶解完全,降温至70℃,加入苯乙基异氰酸酯(0.079g,0.00054mol),保温反应3h。tlc检测反应已完全,停止反应。冷却至室温后加10ml水,抽滤,收集滤饼,烘干,得到的粗品经硅胶柱层析分离(洗脱剂为二氯甲烷:甲醇=50︰1,v/v)得到黄色固体产物0.126g,收率57%。波谱数据:1hnmr(600mhz,dmso-d6):10.09(s,1h),9.99(br.s.,1h),9.35(d,j=2.0hz,1h),8.74(dd,j=1.4,4.7hz,1h),8.66(d,j=5.1hz,1h),8.52(td,j=1.9,7.9hz,1h),7.92~7.95(m,2h),7.88~7.90(m,2h),7.86(s,1h),7.61(dd,j=4.8,7.9hz,1h),7.58(d,j=5.1hz,1h),7.26~7.30(m,2h),7.20~7.23(m,2h),7.17~7.19(m,1h),6.53(br.s.,1h),3.22~3.29(m,2h),2.71(t,j=7.3hz,2h).13cnmr(151mhz,dmso-d6):166.6,162.2,160.3,160.0,159.0,152.1,148.6,144.1,140.0,135.0,132.5,129.1,128.9,128.8,126.5,125.6,124.5,118.2,109.5,41.4,36.4.hrms(+):calcdforc25h24n7o2+[m+h]+454.1986,found454.1981;calcdforc25h23n7o2na+[m+na]+476.1805,found476.1801.
实施例11:4-((4-取代芳基-2-嘧啶基)氨基)苯甲酰肼衍生物的制备:n-苄基-2-(4–((4-(4-氯苯基)-2-嘧啶基)氨基)苯甲酰基)肼-1-硫代甲酰胺
在50ml干燥的双颈瓶里依次加入4-((4-(4-氯苯基)-2-嘧啶基)氨基)苯甲酰肼(0.100g,0.00029mol),n,n-二甲基甲酰胺(7ml),加热至120℃,待固体溶解完全,降温至70℃,加入苄基硫代异氰酸酯(0.048g,0.00032mol),保温反应3.5h。tlc检测反应完全,停止反应。冷却至室温后加10ml水,抽滤,收集滤饼,烘干,得到的粗品经硅胶柱层析分离(洗脱剂为二氯甲烷:甲醇=50︰1,v/v)得到白色固体产物0.110g,收率76%。波谱数据:1hnmr(600mhz,dmso-d6):10.26(s,1h),10.08(s,1h),9.42(s,1h),8.64(d,j=5.3hz,2h),8.20-8.23(m,2h),7.90~7.96(m,4h),7.63~7.67(m,2h),7.52(d,j=5.1hz,1h),7.28~7.34(m,4h),7.19~7.24(m,1h),4.75(d,j=6.0hz,2h).13cnmr(151mhz,dmso-d6):166.2,163.0,160.3,159.9,144.3,140.0,136.3,135.8,129.5,129.2,128.5,127.5,127.0,125.3,118.0,109.2,47.2.hrms(+):calcdforc25h22cln6os+[m+h]+489.1259,found489.1259;calcdforc25h21cln6osna+[m+na]+511.1078,found511.1075。
- 还没有人留言评论。精彩留言会获得点赞!
- 2-甲基-4-氨基-5-氰基嘧啶的环保制备方法
- 一种2-氨基-4,6-二氯-5-甲酰氨基嘧啶的合成方法
- 一种尼洛替尼中间体3-(4-甲基-1h-咪唑-1-基)-5-(三氟甲基)苯胺的制备方法
- 4-氨基-1,2,3-三氮唑[4,5-e]呋咱并[3,4-b]吡嗪-6-氧化物的制作方法
- 氟马替尼的合成方法
- 替格瑞洛中间体4,6-二氯-2-丙巯基-5-氨基嘧啶的制备方法
- 具有催化合成二氢嘧啶酮类化合物功能的对氨基苯磺酸钴的制作方法
- 用于治疗急性、亚急性或慢性咳嗽的二氨基嘧啶p2x3和p2x2/3受体调节剂的制作方法
- 用于治疗急性、亚急性或慢性咳嗽的二氨基嘧啶p2x3和p2x2/3受体调节剂的制作方法
- 一种蛋白激酶抑制剂的合成方法