3R-吲哚甲基-6S-Lys修饰的哌嗪-2,5-二酮,其合成,活性和应用的制作方法
本发明涉及(3r,6s)-3-(lys-氨基正己酰基氨基正丁基)-6-(吲哚-3-甲基)-哌嗪-2,5-二酮。涉及它的制备方法、涉及它的抗肿瘤转移活性,以及涉及它的抗炎活性,因而本发明涉及它在抗肿瘤转移药物和抗炎药物中的应用。本发明属于生物医药领域。
背景技术
肿瘤严重威胁人类的健康。除了自身对肿瘤患者的预后恶劣之外,肿瘤并发的转移进一步恶化患者的预后。例如,超过90%以上肿瘤患者都是死于转移。由于现有抗肿瘤药物没有抗肿瘤转移作用,所以肿瘤化疗的临床疗效不理想。发明抗肿瘤转移的药物是临床的迫切需求。此前,发明人曾公开s,s-,r,r-,r,s-和s,r-四种构型的的二酮哌嗪在0.5μm浓度可抑制hcclm3(高转移人肝癌细胞)迁移和侵袭。后来发明人又公开r,r-构型的二酮哌嗪在5μmol/kg剂量下可抑制c57bl/6小鼠的肿瘤向肺转移。可是最低有效剂量为5μmol/kg。为了降低最低有效剂量,发明人对s,r-构型的二酮哌嗪的氨基正丁基展开了各种修饰。经过3年探索,发现用lys酰化的氨基正己酸酰化s,r-构型的二酮哌嗪的氨基正丁基不仅可使抗肿瘤转移的最低有效剂量降至0.5μmol/kg,而且可使抗炎的最低有效剂量也降至0.5μmol/kg。因为药物的毒副作用都可以随剂量降低而消失,所以有效剂量降低10倍表明了这种结构修饰有突出的技术效果。根据这些发现,发明人提出了本发明。
技术实现要素:
本发明的第一个内容是提供下式的(3r,6s)-3-(lys-氨基正己酰基氨基正丁基)-6-(吲哚-3-甲基)-哌嗪-2,5-二酮。
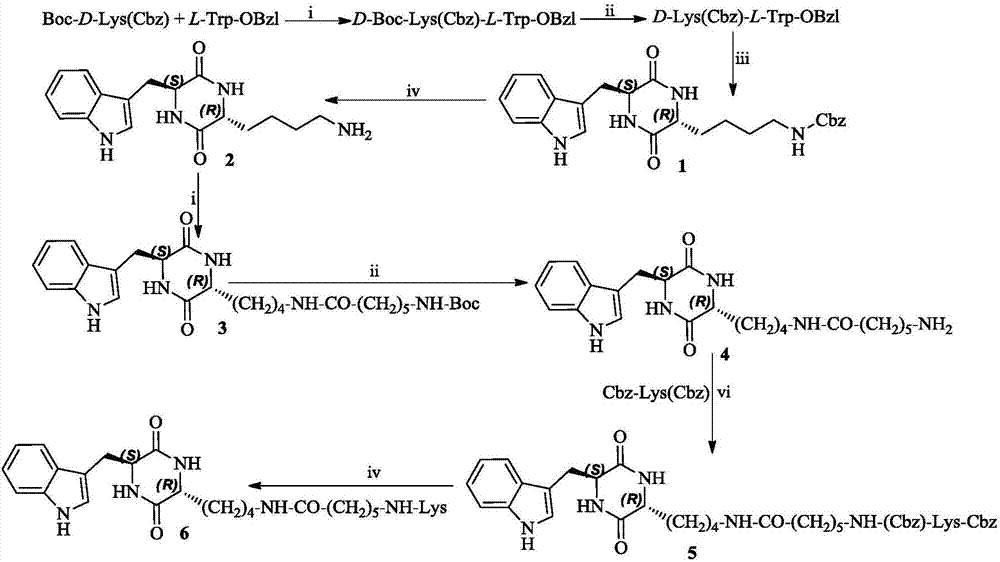
本发明的第二个内容是提供(3r,6s)-3-(lys-氨基正己酰基氨基正丁基)-6-(吲哚-3-甲基)-哌嗪-2,5-二酮的合成方法,该方法包括:
(1)d-boc-lys(cbz)与l-trp-obzl缩合得d-boc-lys(cbz)-d-trp-obzl;
(2)d-boc-lys(cbz)-d-trp-obzl在氯化氢的乙酸乙酯溶液中脱boc得d-lys(cbz)-d-trp-obzl;
(3)d-lys(cbz)-d-trp-obzl在5%碳酸氢钠水溶液饱和的乙酸乙酯溶液中环合得(3r,6s)-3-(苄氧羰基氨基正丁基)-6-(吲哚-3-甲基)-哌嗪-2,5-二酮(1);
(4)化合物1氢解脱苄氧羰基得到(3r,6s)-3-(氨基正丁基)-6-(吲哚-3-甲基)-哌嗪-2,5-二酮(2);
(5)化合物2与boc-氨基正己酸缩合得(3r,6s)-3-(boc-氨基正己酰基氨基正丁基)-6-(吲哚-3-甲基)-哌嗪-2,5-二酮(3);
(6)化合物3在氯化氢的乙酸乙酯溶液中脱boc得到(3r,6s)-3-(氨基正己酰基氨基正丁基)-6-(吲哚-3-甲基)-哌嗪-2,5-二酮(4);
(7)化合物4与cbz-lys(cbz)缩合得(3r,6s)-3-(cbz-lys(cbz)-氨基正己酰基氨基正丁基)-6-(吲哚-3-甲基)-哌嗪-2,5-二酮(5)。
(8)化合物5氢解脱苄氧羰基和苄基得到(3r,6s)-3-(lys-氨基正己酰基氨基正丁基)-6-(吲哚-3-甲基)-哌嗪-2,5-二酮(6)。
本发明的第三个内容是评价(3r,6s)-3-(lys-氨基正己酰基氨基正丁基)-6-(吲哚-3-甲基)-哌嗪-2,5-二酮抑制c57bl/6小鼠抗肺癌转移活性。
本发明的第四个内容是评价(3r,6s)-3-(lys-氨基正己酰基氨基正丁基)-6-(吲哚-3-甲基)-哌嗪-2,5-二酮对icr小鼠炎症的抑制作用。
附图说明
图1(3r,6s)-3-(lys-氨基正己酰基氨基正丁基)-6-(吲哚-3-甲基)-哌嗪-2,5-二酮(6)的合成路线;i)二环己基碳二亚胺(dcc),1-羟基苯并三唑(hobt),n-甲基吗啉(nmm);ii)氯化氢的乙酸乙酯溶液;iii)乙酸乙酯,5%碳酸氢钠水溶液;iv)pd/c,h2。
具体实施方式
为了进一步阐述本发明,下面给出一系列实施例。这些实施例完全是例证性的,它们仅用来对本发明进行具体描述,不应当理解为对本发明的限制。
实施例1制备d-boc-lys(cbz)-l-trp-obzl
冰浴下向1.90g(5.0mmol)d-boc-lys(cbz)与20ml干燥四氢呋喃(thf)的溶液中加入0.68g(5.0mmol)1-羟基苯并三唑(hobt)和1.24g(6.0mmol)二环己基碳二亚胺(dcc)并搅拌30分钟,得到反应液a。将1.47g(5.0mmol)l-trp-obzl溶于20ml干燥thf,加入n-甲基吗啉(nmm)调节ph至9得到反应液b。将反应液b加入反应液a中,室温反应12小时,tlc(二氯甲烷/甲醇,40/1)显示反应完成。反应混合物过滤,滤液减压浓缩,残留物用50ml乙酸乙酯溶解。得到的溶液依次用饱和nahco3水溶液洗(25ml×3),饱和nacl水溶液洗(25ml×3),5%khso4水溶液洗(25ml×3),饱和nacl水溶液洗(25ml×3),饱和nahco3水溶液洗(25ml×3)和饱和nacl水溶液洗(25ml×3)。乙酸乙酯层用无水na2so4干燥12小时。滤除na2so4、滤液减压浓缩,残留物用硅胶柱层析纯化(二氯甲烷/甲醇,40/1),得到2.94g(90%)标题化合物,为无色固体。esi-ms(m/z):657[m+h]+。
实施例2制备d-lys(cbz)-l-trp-obzl
冰浴下向2.62g(4.0mmol)d-boc-lys(cbz)-l-trp-obzl缓慢加入30ml氯化氢的乙酸乙酯溶液(4m)并搅拌4小时,tlc(二氯甲烷/甲醇,40/1)显示反应完成。反应混合物减压浓缩,残留物加30ml无水乙酸乙酯溶解,得到的溶液减压浓缩,残留物加30ml无水乙酸乙酯溶解。该操作重复三次。残留物加30ml无水乙醚借助超声仪使混悬,静置之后去除乙醚,得到2.07g(93%)标题化合物,为黄色粉末。esi-ms(m/z):557[m+h]+。
实施例3制备(3r,6s)-3-(苄氧羰基氨基正丁基)-6-(吲哚-3-甲基)-哌嗪-2,5-二酮(1)
将1.95g(3.5mmol)d-lys(cbz)-l-trp-obzl与50ml乙酸乙酯的溶液用饱和nahco3水溶液充分洗(25ml×3)之后,乙酸乙酯层于80℃搅拌56小时。tlc(二氯甲烷/甲醇,20/1)显示反应完成。反应混合物室温充分放置,过滤,得到0.72g(46%)标题化合物,为无色固体。esi-ms(m/z):449[m+h]+。
实施例4制备(3r,6s)-3-(氨基正丁基)-6-(吲哚-3-甲基)-哌嗪-2,5-二酮(2)
向0.67g(1.5mmol)(3r,6s)-3-(苄氧羰基氨基正丁基)-6-(吲哚-3-甲基)-哌嗪-2,5-二酮(1)与10ml二甲基甲酰胺(dmf)的溶液中加0.07gpd/c(10%),室温通h248小时,tlc(二氯甲烷/甲醇,20/1)显示反应完成。从反应混合物中滤除pd/c,滤液减压浓缩,得到0.48g(95%)标题化合物,为无色固体。esi-ms(m/z):315[m+h]+。
实施例5制备boc-氨基正己酸
搅拌下往0.26g(2.0mmol)氨基正己酸与5ml蒸馏水的溶液中加0.58g(2.6mmol)(boc)2o与5ml二氧六环的溶液。冰浴下将得到的溶液用naoh(2m)的水溶液调ph至9。冰浴下搅拌30分钟后,室温搅拌并用水泵抽气。搅拌期间监测ph并使之始终保持为9,直至tlc(二氯甲烷/甲醇,3/1)显示反应完成。冰浴下,反应混合物用饱和khso4水溶液调节ph至7,减压浓缩。水相用饱和khso4水溶液调节ph至2,用10ml乙酸乙酯充分洗三次,用10ml饱和nacl水溶液洗三次使溶液ph为7,用无水na2so4干燥12小时。过滤除去na2so4,滤液减压浓缩,得到0.41g(89%)标题化合物,为无色固体。esi-ms(m/e):232[m+h]+。
实施例6制备(3r,6s)-3-(boc-氨基正己酰基氨基正丁基)-6-(吲哚-3-甲基)-哌嗪-2,5-二酮(3)
冰浴下向0.28g(1.2mmol)boc-氨基正己酸与5ml无水dmf的溶液中加入0.16g(1.2mmol)hobt和0.29g(1.4mmol)dcc,搅拌30分钟,得到反应液a。将0.38g(1.2mmol)(3r,6s)-3-(氨基正丁基)-6-(吲哚-3-甲基)-哌嗪-2,5-二酮(2)溶于5ml无水dmf,加n-甲基吗啉调节ph至9,得到反应液b。将反应液b加入反应液a中,室温搅拌12小时,tlc(二氯甲烷/甲醇,10/1)显示反应完成。反应混合物过滤,滤液减压浓缩,残留物用硅胶柱层析纯化(二氯甲烷/甲醇,50/1),得到0.12g(19%)标题化合物,为无色固体。esi-ms(m/z):528[m+h]+;1hnmr(300mhz,dmso-d6)δ/ppm=10.903(s,1h),8.046(d,j=1.8hz,1h),7.851(s,1h),7.686(t,j=5.4hz,1h),7.556(d,j=7.8hz,1h),7.314(d,j=7.8hz,1h),7.039(m,2h),6.943(td,j1=7.8hz,j2=0.6hz,1h),6.758(t,j=5.4hz,1h),4.068(m,1h),3.252(dd,j1=14.4hz,j2=4.2hz,1h),2.924(m,6h),1.995(t,j=7.2hz,2h),1.292(m,21h)。
实施例7制备(3r,6s)-3-(氨基正己酰基氨基正丁基)-6-(吲哚-3-甲基)-哌嗪-2,5-二酮(4)
采用实施例2的方法从0.11g(0.2mmol)(3r,6s)-3-(boc-氨基正己酰基氨基正丁基)-6-(吲哚-3-甲基)-哌嗪-2,5-二酮(3)得到0.08g(96%)标题化合物,为无色固体。esi-ms(m/z):428[m+h]+;1hnmr(300mhz,dmso-d6)δ/ppm=10.924(s,1h),8.071(s,1h),7.866(s,1h),7.729(t,j=5.4hz,1h),7.568(d,j=7.5hz,1h),7.321(d,j=7.5hz,1h),7.044(m,2h),6.949(t,j=7.5hz,1h),4.070(m,1h),3.239(dd,j1=14.4hz,j2=3.6hz,1h),2.991(m,4h),2.694(m,2h),2.021(t,j=7.5hz,2h),1.467(m,6h),1.240(m,6h)。
实施例8制备(3r,6s)-3-(cbz-lys(cbz)-氨基正己酰基氨基正丁基)-6-(吲哚-3-甲基)-哌嗪-2,5-二酮(5)
采用实施例6的方法从0.07g(0.18mmol)cbz-lys(cbz)和0.08g(0.18mmol)(3r,6s)-3-(氨基正己酰基氨基正丁基)-6-(吲哚-3-甲基)-哌嗪-2,5-二酮(4)得到0.03g(17%)标题化合物,为无色固体。esi-ms(m/z):824[m+h]+;1hnmr(300mhz,dmso-d6)δ/ppm=10.898(s,1h),8.037(d,j=2.4hz,1h),7.832(m,2h),7.680(t,j=5.4hz,1h),7.568(d,j=7.8hz,1h),7.337(m,1h),7.227(t,j=5.4hz,1h),7.014(m,2h),6.921(td,j1=7.8hz,j2=0.9hz,1h),5.012(s,2h),4.998(s,2h),4.071(m,1h),3.892(m,1h),3.555(m,1h),3.238(dd,j1=14.4hz,j2=4.2hz,1h),3.006(m,6h),2.277(m,1h),1.995(t,j=7.2hz,2h),1.350(m,18h)。
实施例9制备(3r,6s)-3-(lys-氨基正己酰基氨基正丁基)-6-(吲哚-3-甲基)-哌嗪-2,5-二酮(6)
按照实施例4的方法从0.03g(0.03mmol)(3r,6s)-3-(cbz-lys(cbz)-氨基正己酰基氨基正丁基)-6-(吲哚-3-甲基)-哌嗪-2,5-二酮(5c)得到0.02g(93%)标题化合物,为无色固体。esi-ms(m/z):556[m+h]+;mp160-162℃;
实施例10测定化合物6的抗肿瘤转移活性
本测定模型用lewis小鼠肺癌细胞(llc,购自atcc)接种,选用dmem培养基(含10%经灭活的胎牛血清,1×105u/l青霉素和100mg/l链霉素),按照贴壁细胞培养方法每两天传代一次,富集细胞。待细胞生长状态良好并处于对数生长期时消化细胞,用生理盐水调整细胞密度至1×107个/ml。胎盘蓝染色,使活细胞计数>95%。取近交系c57bl/6雄性小鼠(spf级,体重20±2g),左手固定小鼠。用75%乙醇对小鼠右前肢腋窝皮肤消毒。右手持1ml无菌注射器往小鼠腋部皮下注射llc肿瘤细胞悬液,每只小鼠注射0.2ml。小鼠接种10天后,长出直径约4-5mm的肿瘤即为瘤源。接种10天的lewis肺癌荷瘤小鼠乙醚麻醉,脱颈椎处死。用75%乙醇浸泡10min,消毒,在超净工作台上剥离瘤体。选择生长良好的肿瘤组织在无菌平皿中剪碎,置于玻璃制造的组织匀浆器内。按瘤块重比生理盐水体积为1比3(g比ml)的比例加温度为4℃的生理盐水,轻轻研磨制成细胞悬液。细胞悬液过200目细胞筛制单细胞悬液。用生理盐水调单细胞悬液的细胞密度至1.5×107个/ml。胎盘蓝染色,使活细胞计数>95%。左手固定近交系c57bl/6雄性小鼠,用75%的乙醇对小鼠右前肢腋窝皮肤消毒。右手持1ml无菌注射器于小鼠腋部皮下注射瘤细胞悬液,每只注射0.2ml。接种10天后小鼠长出直径4-5mm的肿瘤,按测得的肿瘤体积将接种小鼠随机分组。每组12只小鼠。接种肿瘤的第11天小鼠或口服公认的抗肿瘤转移肽rgds的生理盐水溶液(剂量为20μmol/kg/天)或口服化合物6的生理盐水溶液(剂量为0.5μmol/kg/天)或口服化合物4的生理盐水溶液(剂量为5μmol/kg/天)或口服生理盐水(剂量为10ml/kg/天),每天给1次药,连续给药12天。最后一次给药的次日乙醚麻醉脱颈椎处死,取小鼠的肺并计算肿瘤肺部转移的瘤节数。用t检验对数据进行统计分析。结果见表1。在0.5μmol/kg剂量下化合物6不仅有效地抑制肿瘤肺转移,而且活性与剂量比它们高40倍的rgds及剂量比它们高10倍的化合物4没有显著性差异。这些数据表明,本发明有显著的技术效果。
表1化合物6的抗肿瘤转移活性
a)与生理盐水比p<0.01,与rgds及化合物4比p>0.05;n=12.
实施例11测定化合物6的抗炎活性
因为二甲苯引起的小鼠耳肿胀被公认为急性炎症模型,所以本发明在二甲苯引起的小鼠耳肿胀模型上测定化合物6的治疗作用。因为阿司匹林是治疗急性炎症的阳性药,所以本发明选择阿司匹林为阳性对照药。icr雄性小鼠(spf级,体重20±2g)在温度为22℃的环境静息2天,自由饮水和进食。之后,随机分为生理盐水组(剂量为0.2ml/只),阿司匹林组(剂量为1.11mmol/kg),化合物4组(剂量为5μmol/kg)及化合物6组(剂量为0.5μmol/kg),每组12只小鼠。测定时小鼠按所在组或口服生理盐水,或口服阿司匹林,或口服化合物4,或口服化合物6。给药30min后,往小鼠的左耳廓均匀涂抹30μl二甲苯,2h后小鼠接受乙醚麻醉,断颈处死,剪下左右两耳,用7mm的打孔器在两耳的相同位置取圆形耳片,称重,求出两耳肿胀差值作为肿胀度。即肿胀度=左耳圆片重量–右耳圆片重量。结果见表2。在0.5μmol/kg剂量下化合物6不仅可以有效地抑制二甲苯引起的小鼠耳肿胀,而且活性与剂量比它们高2220倍的阿司匹林及剂量比它们高10倍的化合物4没有显著性差异。这些数据表明,本发明有显著的技术效果。
表2化合物6对二甲苯引起的小鼠耳肿胀的影响
a)与生理盐水比p<0.01,与阿司匹林及化合物4比p>0.05;n=12。
- 还没有人留言评论。精彩留言会获得点赞!