巴瑞替尼的杂质及其制备、检测方法与流程
本发明涉及医药化工领域,具体涉及巴瑞替尼的杂质、其制备方法、检测方法以及含有所述杂质的组合物。
背景技术:
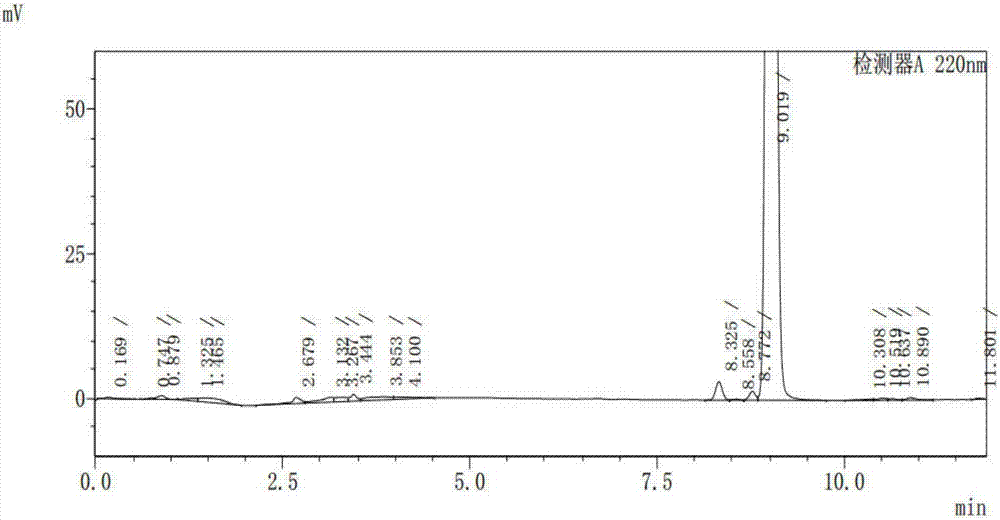
巴瑞替尼(baricitinib,olumiant,i)是由礼来制药与其合作伙伴incyte公司共同开发的一种选择性jak1和jak2抑制剂,能抑制il-6和il-23等多种炎性细胞因子的细胞内信号传导,化学名为1-(乙基磺酰基)-3-[4-(7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基]-3-氮杂环丁烷乙腈,其结构式如式(i)所示。该品已于2017年被欧盟批准用于治疗中至重度类风湿性关节炎、对目前的风湿性关节炎药物不反应或者不耐受的成年人,其可作为单一药物使用,也可与广泛使用的甲氨蝶呤一起使用。现有专利文献wo2009114512a1、cn105294699a、cn105541891a、wo2016088094a1、wo2016125080a2、wo2016205487a1中公开了若干种巴瑞替尼的制备方法,但对于可能的杂质情况,文献未有涉及。发明人在工艺路线选择研究实验过程中发现,不管是依据以上专利文本中公开的制备方法,还是依据自研工艺,在最终的巴瑞替尼产品中均会存在至少三种杂质,且其产生很难通过工艺的优化选择而避免。因此,确定巴瑞替尼杂质的化学结构以及制备方法,对后续的制剂稳定性研究、建立检测方法、分析杂质含量并确定合理的杂质限度,以及临床用药安全检测均起到了至关重要的作用。技术实现要素:本发明提供了巴瑞替尼的杂质,选自下列化合物中的一种或多种:如下所示化合物a、化合物b、化合物c本发明还提供上述杂质a、杂质b和杂质c的制备方法,其合成工艺分别如下:杂质a:具体包括以下步骤:在碱存在下,巴瑞替尼在溶剂中由过氧化氢氧化水解得到化合物a。其中所述碱包括但不限于碳酸钾、氢氧化钾、氢氧化钠、碳酸钠中的一种或几种;巴瑞替尼与所述碱的用量摩尔比为1:(1.5~2.5)。其中过氧化氢采用重量百分比为30%~100%的过氧化氢溶液;巴瑞替尼与所述过氧化氢的用量摩尔比为1:(10~15)。其中溶剂包括但不限于dmso、甲醇、乙醇、水中的一种或几种。反应温度优选室温至50℃。杂质b:具体包括以下步骤:在有溶剂或无溶剂状态下,巴瑞替尼在酸性条件下水解得到化合物b。其中所述溶剂包括但不限于甲醇、二氧六环、dmf、dmso中的一种或几种。其中所述酸包括但不限于浓盐酸、浓硫酸、浓磷酸中的一种或几种。反应温度优选60℃至溶剂回流温度。杂质c:具体包括以下步骤:新戊酸[4-(1h-吡唑-4-基)-7h-吡咯并[2,3-d]嘧啶-7-基]甲酯和3-(氰基亚甲基)氮杂环丁烷-1-甲酸叔丁酯在dbu作用下进行michael加成反应得到中间体c1;中间体c1在碱性条件下脱除特戊酰氧基甲基保护得到中间体c2;中间体c2再在酸性条件下脱除boc保护基并碱化得目标化合物c。其中,新戊酸[4-(1h-吡唑-4-基)-7h-吡咯并[2,3-d]嘧啶-7-基]甲酯与3-(氰基亚甲基)氮杂环丁烷-1-甲酸叔丁酯的投料当量比为1:(1~1.2),反应温度为室温。所述中间体c1脱保护所采用的碱选自氢氧化钠、氢氧化钾、氢氧化锂、碳酸钠、碳酸钾中的一种或几种。所述中间体c2脱保护所采用的酸选自盐酸水溶液,重量比为10-20%。本发明还提供一种药物组合物,其中组合物中含有巴瑞替尼以及巴瑞替尼杂质化合物a、b、c,其中各杂质的含量均不高于0.15%,更为优选是不高于0.1%。本发明还提供一种检测前述巴瑞替尼杂质a、杂质b或杂质c的hplc分析方法,其特征在于,检测条件如下:仪器设备:高效液相色谱仪,色谱柱:agilentzorbaxsb-aq(4.6×250mm,5μm)色谱条件:流动相a:磷酸盐缓冲液;流动相b:乙腈;检测波长:220nm;柱温:40℃;流速:1.0ml/min;进样量:5μl,梯度洗脱,洗脱程序如下:时间(min)流动相a(%)流动相b(%)09010107030207030304060454060509010559010磷酸盐缓冲液配制:取磷酸二氢钾5.4425g,加水2l,溶解后,用磷酸调ph=2.2,过滤,脱气,即得。样品溶液配制:流动相a:流动相b=1:1的溶液做溶剂,配制成含化合物a或b或c0.2mg/ml的样品溶液。通过本发明可以为巴瑞替尼的质量控制和临床用药安全检测提供标准对照品,保证临床用药的安全可靠性。并且,上述各杂质的制备方法操作简单,反应条件温和,能以较高收率获得高纯度的各杂质化合物。并且,上述化合物a、化合物b和化合物c可以作为杂质对照品控制巴瑞替尼原料和制剂的纯度。附图说明附图1:实施例1化合物a的hplc图谱;附图2:实施例1化合物a的1hnmr图谱;附图3:实施例3化合物b的hplc图谱;附图4:实施例3化合物b的1hnmr图谱;附图5:实施例5化合物c的hplc图谱;附图6:实施例5化合物c的1hnmr图谱。具体实施方式本发明将于下文通过实施例更加详细的描述,这些实施例示例性地用于进一步说明,且不应当视为对本发明的限制。本发明中的hplc条件如下:仪器设备:高效液相色谱仪,色谱柱:agilentzorbaxsb-aq(4.6×250mm,5μm)色谱条件:流动相a:磷酸盐缓冲液;流动相b:乙腈;检测波长:220nm;柱温:40℃;流速:1.0ml/min;进样量:5μl,梯度洗脱;洗脱程序如下:时间(min)流动相a(%)流动相b(%)09010107030207030304060454060509010559010磷酸盐缓冲液配制:取磷酸二氢钾5.4425g,加水2l,溶解后,用磷酸调ph=2.2,过滤,脱气,即得。样品溶液配制:流动相a:流动相b=1:1的溶液做溶剂,配制成含化合物a或b或c0.2mg/ml的样品溶液。实施例1杂质a的制备称取巴瑞替尼(1.0g,2.7mmol)投入到100ml反应瓶中,加入20mldmso,搅拌溶解,加入碳酸钾(0.6g,4.3mmol),冷却至10℃以下,慢慢滴入4.0ml30%双氧水,滴完后自然升温至室温,搅拌10min左右,tlc确认反应完全后,加入100ml水,乙酸乙酯萃取(30ml×3),合并有机相,水洗(30ml×2),饱和食盐水洗涤,无水硫酸钠干燥30min,过滤,滤液减压浓缩至干,得到白色固体产物500mg,产率47.7%,hplc纯度96.96%。ms-esi(m+1):m/z391.1;1hnmr(500mhz,d6-dmso)δ:12.07(s,1h),10.17(s,1h),8.68(d,2h),8.40(s,1h),7.58(d,1h),7.47(d,1h),6.98(d,2h),4.53(d,2h),4.37(d,2h),3.30(s,2h),3.15-3.22(m,2h),3.00(s,2h),1.24(t,3h).实施例2杂质a的制备称取巴瑞替尼(1.0g,2.7mmol)投入到50ml反应瓶中,加入15ml甲醇和1mldmso,然后再在室温下加入6ml的1mol/lnaoh溶液和2mlh2o2。反应混合物于50℃下加热3h,tlc确认反应完全后,冷却至室温,加入100ml水,乙酸乙酯萃取(30ml×3),合并有机相,水洗(30ml×2),饱和食盐水洗涤,无水硫酸钠干燥30min,过滤,滤液减压浓缩至干,得到白色固体产物612mg,产率58.4%,hplc纯度95.98%。实施例3杂质b的制备称取巴瑞替尼(2.0g,5.4mmol)投入到100ml反应瓶中,慢慢加入30ml浓盐酸,升温至100℃,反应1~2h,tlc确认反应完全后,加入30ml水,冷却至0℃,用30%的naoh溶液调ph至ph9~10左右,乙酸乙酯萃取(50ml×3),合并有机层,水洗(50ml×2),无水硫酸钠干燥30min,过滤,滤液于45℃下减压浓缩至干,得到类白色固体800mg,产率38.1%,hplc纯度94.67%。ms-esi(m+1):m/z391.11hnmr(300mhz,d6-dmso)δ:13.19(brs,1h),9.20(s,1h),8.94(s,1h),8.75(s,1h),7.96(s,1h),7.61(t,1h),7.44(s,1h),4.85(d,1h),4.69(d,1h),3.68(s,2h),3.27-3.51(m,2h),2.95(q,2h),1.03-1.15(m,3h).实施例4杂质b的制备称取巴瑞替尼(8.0g,21.5mmol)投入到500ml反应瓶中,慢慢加入120ml浓盐酸,升温至100℃,反应1~1h,tlc确认反应完全后,加入120ml水,冷却至0℃,用30%的naoh溶液调ph至ph9~10左右,乙酸乙酯萃取(100ml×3),合并有机层,水洗(100ml×2),无水硫酸钠干燥30min,过滤,滤液于45℃下减压浓缩至干,得类白色固体3.612g,产率43.0%,hplc纯度93.89%。实施例5杂质c的制备(1)称取新戊酸[4-(1h-吡唑-4-基)-7h-吡咯并[2,3-d]嘧啶-7-基]甲酯(10.0g,33.4mmol)和3-(氰基亚甲基)氮杂环丁烷-1-甲酸叔丁酯(6.8g,35.07mmol,1.05eq.)投入到100ml反应瓶中,加入30mldmf,搅拌固体未全溶,室温下滴加dbu(0.25g,1.67mmol,0.05eq.),固体慢慢溶解后又迅速析出固体产物,继续搅拌反应1~2h,tlc确认反应完全后,加入150ml水,搅拌30min,过滤,滤饼水洗(20ml×4),所得固体于60℃下真空干燥10h,得到类白色固体产物16.3g。(2)称取步骤(1)中所述产物(8.0g,16.2mmol)投入到500ml反应瓶中,加入40ml甲醇和160ml四氢呋喃,搅拌溶解,室温下滴入18.2ml1mol/l氢氧化钠水溶液,室温搅拌3h,tlc确认反应完全,冷却至10℃以下,用1mol/lhcl溶液调ph至ph7~7.5左右,然后于45℃下减压浓缩除去甲醇和四氢呋喃,向剩余物料中加入50ml水,过滤,滤饼用水洗至ph中性,于50℃下真空干燥10h,得到白色固体产物4.6g。(3)将步骤(2)所得产物(4.6g,12.1mmol)投入到250ml反应瓶中,加入64.4ml乙腈和64.4ml3mol/lhcl溶液,室温搅拌18h,有固体析出,直接过滤,滤饼用乙腈洗涤一次,所得产物于50℃下真空干燥10h,得到白色固体。将所得固体溶于30ml水,采用1mol/lnaoh溶液碱化至溶液ph9~10左右,析出固体过滤,滤饼水洗至ph中性,于50℃下真空干燥10h,得白色固体产物2.4g。三步总产率25.7%,hplc纯度98.66%。ms-esi(m+1):m/z280.11hnmr(500mhz,d6-dmso)δ:13.18(s,1h),10.15(s,1h),9.92(s,1h),9.44(s,1h),8.92(d,2h),7.98(s,1h),7.47(s,1h),4.73(d,2h),4.43(d,2h),3.99(s,1h).当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1