一种β‑酰基‑烯丙基甲硫醚衍生物及其制备方法与流程
本发明属于有机硫化合物及其制备方法技术领域,具体涉及一种β-酰基-烯丙基甲硫醚衍生物及其制备方法。
背景技术:
有机硫化合物作为仅次于含有氮和氧的有机化合物被广泛应用于有机药物合成和生物化学。大多数有机硫化合物在生物学作用研究中都具有抑癌和杀菌作用。据统计,2012年美国销售的最好的药物中约20%是有机硫化合物的药物。传统的有机硫化合物的制备都需要用到具有刺激性味道的硫醇作为原料,在碱作用下与卤代烃反应来制备有机硫醚,或者通过过渡金属如钯或者铑作为催化剂与炔烃或者联烯制备烯丙基硫醚(ref.angew.chem.int.ed.2015,54,3121;angew.chem.int.ed.2015,54,15818)。烯丙基硫醚类化合物既具有很好的生物活性,也是有机合成中非常重要的中间体(ref.org.lett.2016,18,6042-6045),此外官能团化的β-酰基-烯丙基硫醚是生物偶联试剂的前体化合物,在人工合成具有生物活性的蛋白质和多肽中有着非常重要的应用价值(ref.chem.eur.j.2011,17,9697-9707)。而传统的制备β-酰基-烯丙基硫醚类化合物通常需要三到四步合成才能实现。而且要使用刺激性气味的硫醇或昂贵的过渡金属催化剂。
技术实现要素:
本发明的目的是为了解决传统β-酰基-烯丙基硫醚类化合物的制备存在制备步骤繁琐、原料价格昂贵和环境不友好的技术问题,提供一种β-酰基-烯丙基甲硫醚衍生物及其制备方法。
为解决上述技术问题,本发明采用的技术方案为:
一种β-酰基-烯丙基甲硫醚衍生物,其化学结构式为:
所述ar为苯基、呋喃、噻吩、萘环、3-甲氧基苯基、4-硝基苯基、4-联苯基、3-溴苯基、3-吡啶基、5-溴噻吩基、苯并呋喃。
一种所述2-((甲硫基)甲基)-1-苯基丙-2-烯-1-酮衍生物的制备方法,化学反应方程式为:
所述ar为苯基、呋喃、噻吩、萘环、3-甲氧基苯基、4-硝基苯基、4-联苯基、3-溴苯基、3-吡啶基、5-溴噻吩基、苯并呋喃。
制备方法包括以下步骤:
1)将芳基甲基酮、dmso、碱金属盐、四氢吡咯烷、对甲苯磺酸-水合物按照1:28:0.8:1:1的摩尔比放入反应容器中,以无水醋酸作为溶剂,在室温下混合均匀,随后在150℃条件下反应2小时;
2)反应完成后将反应物冷却至室温,用乙酸乙酯溶解,并依次用水、饱和氯化钠水溶液洗涤,有机相经无水硫酸钠干燥后,过滤,旋去溶剂;
3)将旋去溶剂后的反应物用硅胶柱层析分离纯化,旋蒸除去溶剂,油泵抽干,既得目标产物。
所述芳基甲基酮为苯乙酮、呋喃甲基酮、噻吩甲基酮、2-萘甲基酮、3-甲氧基苯基甲基酮、4-硝基苯基甲基酮、4-联苯基甲基酮、3-溴苯基甲基酮、3-吡啶基甲基酮、5-溴噻吩基甲基酮、苯并呋喃甲基酮中的任一种。
所述碱金属盐为无水醋酸钠、醋酸锂、醋酸钾、醋酸铯或氟化钠中的任一种。
用核磁共振氢谱(1hnmr)、碳谱(13cnmr)以及高分辨质谱证实了β-酰基-烯丙基甲硫醚衍生物的结构(如附图1-22)。检测所用仪器为:avanceiiihd600mhz型核磁共振仪,其中氘代氯仿为内标(氢谱,氘代氯仿:δ7.26ppm).(碳谱,氘代氯仿:δ77ppm)。thermoscientificqexactive型高分辨质谱仪。
本发明采用以上技术方案,与现有技术相比,本发明的优点具体体现为:
(1)首次采用苯乙酮衍生物与dmso反应,相对于以往的硫醚的合成方法,该方法不仅简单环保,而且产物中还包含有α,β不饱和羰基的结构单元,作为有机合成底物,能很好的通过多种转化反应进一步形成更有价值的化合物;
(2)传统制备硫醚的方法通常需要具有刺激性气味的硫醇做为原料经直接亲核取代或过渡金属催化的c-x/s-h偶联反应得到。而该方法在无需金属催化剂的条件下就可以得到产物,同时以廉价易得,低毒的二甲基亚砜做为含硫化合物的来源;
(3)本发明提供了一个快速直接产生有机合成中非常重要的中间体β-酰基烯丙基硫醚的新途径,其原料为简单易得的甲基酮和二甲基亚砜,且不涉及任何氧化剂和过渡金属催化剂的使用。降低了反应成本且大大减少了对环境的污染。该设计方案不仅可以给工业上处理二甲基亚砜提供新的途径,而且所转化生成的β-酰基烯丙基甲硫醚产物在医药,农药,以及功能材料等领域将具有非常广泛的应用前景;
(4)本发明所述反应底物的官能团适用范围较广,包括:含卤素、烷基、烷氧基等官能团底物,以及包含有吡啶、呋喃、噻吩、萘环、联苯等结构的底物。
附图说明
图1为2-((甲硫基)甲基)-1-苯基丙-2-烯-1-酮的氢谱;
图2为2-((甲硫基)甲基)-1-苯基丙-2-烯-1-酮的碳谱;
图3为1-(呋喃-2-基)-2-((甲硫基)甲基)丙-2-烯-1-酮的氢谱;
图4为1-(呋喃-2-基)-2-((甲硫基)甲基)丙-2-烯-1-酮的碳谱;
图5为2-(甲硫基)甲基)-1-(噻吩-2-基)丙-2-烯-1-酮的氢谱;
图6为2-(甲硫基)甲基)-1-(噻吩-2-基)丙-2-烯-1-酮的碳谱;
图7为2-((甲硫基)甲基)-1-(萘-2-基)丙-2-烯-1-酮的氢谱;
图8为2-((甲硫基)甲基)-1-(萘-2-基)丙-2-烯-1-酮的碳谱;
图9为1-(3-甲氧基苯基)-2-((甲硫基)甲基)丙-2-烯-1-酮的氢谱;
图10为1-(3-甲氧基苯基)-2-((甲硫基)甲基)丙-2-烯-1-酮的碳谱;
图11为2-((甲硫基)甲基)-1-(4-硝基苯基)丙-2-烯-1-酮的氢谱;
图12为2-((甲硫基)甲基)-1-(4-硝基苯基)丙-2-烯-1-酮的碳谱;
图13为1-([1,1'-联苯]-4-基)-2-((甲硫基)甲基)丙-2-烯-1-酮的氢谱;
图14为1-([1,1'-联苯]-4-基)-2-((甲硫基)甲基)丙-2-烯-1-酮的碳谱;
图15为1-(3-溴苯基)-2-((甲硫基)甲基)丙-2-烯-1-酮的氢谱;
图16为1-(3-溴苯基)-2-((甲硫基)甲基)丙-2-烯-1-酮的碳谱;
图17为2-((甲硫基)甲基)-1-(吡啶-3-基)丙-2-烯-1-酮的氢谱;
图18为2-((甲硫基)甲基)-1-(吡啶-3-基)丙-2-烯-1-酮的碳谱;
图19为1-(5-溴噻吩-2-基)-2-((甲硫基)甲基)丙-2-烯-1-酮的氢谱;
图20为1-(5-溴噻吩-2-基)-2-((甲硫基)甲基)丙-2-烯-1-酮的碳谱;
图21为1-(苯并呋喃-2-基)-2-((甲硫基)甲基)丙-2-烯-1-酮的氢谱;
图22为1-(苯并呋喃-2-基)-2-((甲硫基)甲基)丙-2-烯-1-酮的碳谱。
具体实施方式
实施例1
本实施例中的β-酰基-烯丙基甲硫醚衍生物为2-((甲硫基)甲基)-1-苯基丙-2-烯-1-酮。
其制备方法包括以下步骤:
1)将苯乙酮(0.0292ml,0.25mmol)、dmso(0.5ml)、无水醋酸钠(0.0164g,0.2mmol)、四氢吡咯烷(0.0209ml,0.25mmol)、对甲苯磺酸-水合物(0.0476g,0.25mmol)和无水醋酸(2ml)放入干净干燥的密闭反应容器中,在室温下混合均匀,随后在150℃条件下反应2小时;
2)反应完成后将反应物冷却至室温,用40ml乙酸乙酯将反应体系稀释溶解,加入20ml水,摇晃,静置,移除下面的水相后,再加入20ml饱和食盐水,摇晃,静置后,移除下面水相,有机相经无水硫酸钠干燥后,过滤,减压旋去溶剂;
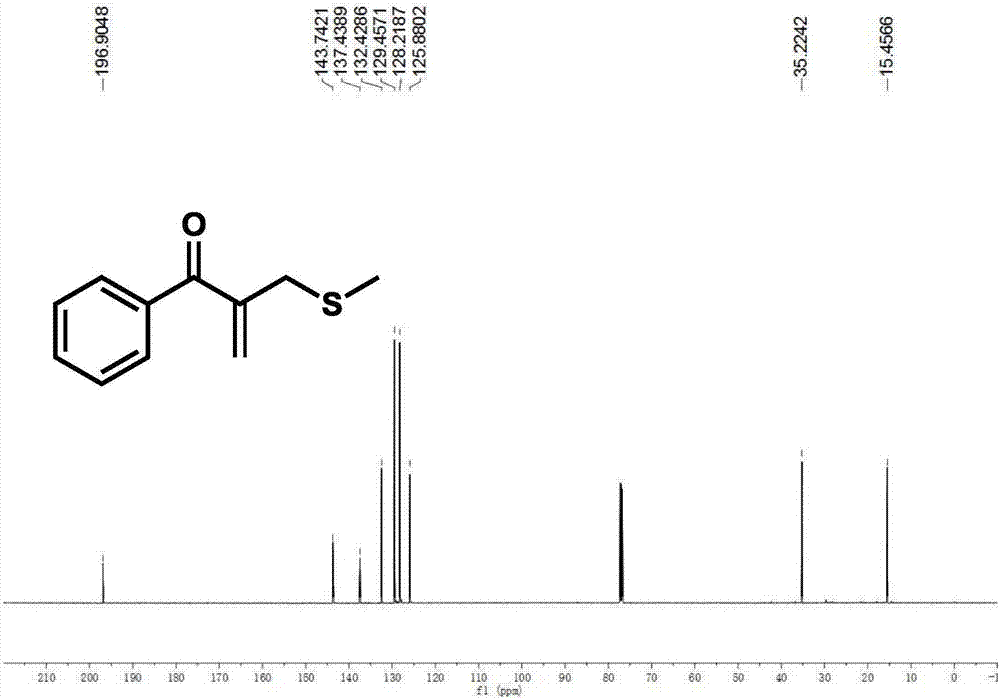
3)将旋去溶剂后的反应物用硅胶柱层析(石油醚/乙酸乙酯=10:1,v/v)分离纯化,旋蒸除去溶剂,油泵抽干,既得黄色油状目标产物39g,产率81%。如图1和2所示,1hnmr(600mhz,cdcl3)δ7.76(d,j=7.7hz,2h),7.55(t,j=7.2hz,1h),7.44(t,j=7.4hz,2h),5.91(s,1h),5.67(s,1h),3.53(s,2h),2.12(s,3h);13cnmr(151mhz,cdcl3)δ196.90,143.74,137.44,132.43,129.46,128.22,125.88,35.22,15.46.hrms(esi+):计算值c11h13os[m+h]+193.0692,实测值193.0683。
实施例2
本实施例中的β-酰基-烯丙基甲硫醚衍生物为1-(呋喃-2-基)-2-((甲硫基)甲基)丙-2-烯-1-酮。
其制备方法包括以下步骤:
1)将2-乙酰基呋喃(0.0251ml,0.25mmol)、dmso(0.5ml)、无水醋酸钠(0.0164g,0.2mmol)、四氢吡咯烷(0.0209ml,0.25mmol)、对甲苯磺酸-水合物(0.0476g,0.25mmol)和无水醋酸(2ml)放入干净干燥的密闭反应容器中,在室温下混合均匀,随后在150℃条件下反应2小时;
2)反应完成后将反应物冷却至室温,用40ml乙酸乙酯将反应体系稀释溶解,加入20ml水,摇晃,静置,移除下面的水相后,再加入20ml饱和食盐水,摇晃,静置后,移除下面水相,有机相经无水硫酸钠干燥后,过滤,减压旋去溶剂;
3)将旋去溶剂后的反应物用硅胶柱层析(石油醚/乙酸乙酯=5:1,v/v)分离纯化,旋蒸除去溶剂,油泵抽干,既得黄色油状目标产物28.1g,产率62%。如图3和4所示,1hnmr(600mhz,cdcl3)δ7.64(d,j=0.6hz,1h),7.18(d,j=3.5hz,1h),6.54(dd,j=3.5,1.6hz,1h),6.03(s,1h),5.84(s,1h),3.50(s,2h),2.06(s,3h);13cnmr(151mhz,cdcl3)δ182.71,152.03,147.12,143.43,124.34,119.96,112.04,35.20,15.23.hrms(esi+):计算值c9h11o2s[m+h]+183.0474,实测值183.0474。
实施例3
本实施例中的β-酰基-烯丙基甲硫醚衍生物为2-(甲硫基)甲基)-1-(噻吩-2-基)丙-2-烯-1-酮。
其制备方法包括以下步骤:
1)将2-乙酰基噻吩(0.0270ml,0.25mmol)、dmso(0.5ml)、无水醋酸钠(0.0164g,0.2mmol)、四氢吡咯烷(0.0209ml,0.25mmol)、对甲苯磺酸-水合物(0.0476g,0.25mmol)和无水醋酸(2ml)放入干净干燥的密闭反应容器中,在室温下混合均匀,随后在150℃条件下反应2小时;
2)反应完成后将反应物冷却至室温,用40ml乙酸乙酯将反应体系稀释溶解,加入20ml水,摇晃,静置,移除下面的水相后,再加入20ml饱和食盐水,摇晃,静置后,移除下面水相,有机相经无水硫酸钠干燥后,过滤,减压旋去溶剂;
3)将旋去溶剂后的反应物用硅胶柱层析(石油醚/乙酸乙酯=5:1,v/v)分离纯化,旋蒸除去溶剂,油泵抽干,既得黄色油状目标产物36g,产率73%。如图5和6所示,1hnmr(600mhz,cdcl3)δ7.65(ddd,j=6.0,4.3,1.0hz,2h),7.09(dd,j=4.9,3.9hz,1h),5.82(s,1h),5.77(s,1h),3.47(s,2h),2.05(s,3h);13cnmr(151mhz,cdcl3)δ188.28,144.13,143.33,134.29,133.93,127.89,123.40,35.59,15.28.hrms(esi+):计算值c9h11os2[m+h]+199.0246,实测值199.0245。
实施例4
本实施例中的β-酰基-烯丙基甲硫醚衍生物为2-((甲硫基)甲基)-1-(萘-2-基)丙-2-烯-1-酮。
其制备方法包括以下步骤:
1)将2-萘乙酮(0.0426g,0.25mmol)、dmso(0.5ml)、无水醋酸钠(0.0164g,0.2mmol)、四氢吡咯烷(0.0209ml,0.25mmol)、对甲苯磺酸-水合物(0.0476g,0.25mmol)和无水醋酸(2ml)放入干净干燥的密闭反应容器中,在室温下混合均匀,随后在150℃条件下反应2小时;
2)反应完成后将反应物冷却至室温,用40ml乙酸乙酯将反应体系稀释溶解,加入20ml水,摇晃,静置,移除下面的水相后,再加入20ml饱和食盐水,摇晃,静置后,移除下面水相,有机相经无水硫酸钠干燥后,过滤,减压旋去溶剂;
3)将旋去溶剂后的反应物用硅胶柱层析(石油醚/乙酸乙酯=5:1,v/v)分离纯化,旋蒸除去溶剂,油泵抽干,既得黄色油状目标产物47.1g,产率78%。如图7和8所示,1hnmr(600mhz,cdcl3)δ8.28(s,1h),7.98–7.81(m,4h),7.57(dt,j=14.9,7.1hz,2h),5.96(s,1h),5.74(s,1h),3.60(s,2h),2.17(s,3h);13cnmr(151mhz,cdcl3)δ196.59,143.68,134.99,134.39,131.91,130.87,129.10,127.99,127.98,127.47,126.48,125.45,124.99,35.18,15.32.hrms(esi+):计算值c15h15os[m+h]+243.0838,实测值243.0838。
实施例5
本实施例中的β-酰基-烯丙基甲硫醚衍生物为1-(3-甲氧基苯基)-2-((甲硫基)甲基)丙-2-烯-1-酮。
其制备方法包括以下步骤:
1)将3-甲氧基苯乙酮(0.0343ml,0.25mmol)、dmso(0.5ml)、无水醋酸钠(0.0164g,0.2mmol)、四氢吡咯烷(0.0209ml,0.25mmol)、对甲苯磺酸-水合物(0.0476g,0.25mmol)和无水醋酸(2ml)放入干净干燥的密闭反应容器中,在室温下混合均匀,随后在150℃条件下反应2小时;
2)反应完成后将反应物冷却至室温,用40ml乙酸乙酯将反应体系稀释溶解,加入20ml水,摇晃,静置,移除下面的水相后,再加入20ml饱和食盐水,摇晃,静置后,移除下面水相,有机相经无水硫酸钠干燥后,过滤,减压旋去溶剂;
3)将旋去溶剂后的反应物用硅胶柱层析(石油醚/乙酸乙酯=5:1,v/v)分离纯化,旋蒸除去溶剂,油泵抽干,既得黄色油状目标产物39.5g,产率71%。如图9和10所示,1hnmr(600mhz,cdcl3)δ7.38–7.28(m,3h),7.08(d,j=7.1hz,1h),5.89(s,1h),5.68(s,1h),3.83(s,3h),3.51(s,2h),2.11(s,3h);13cnmr(151mhz,cdcl3)δ196.58,159.43,143.66,138.67,129.13,125.77,122.10,118.72,113.84,55.31,35.23,15.38.hrms(esi+):计算值c12h15o2s[m+h]+223.0787,实测值223.0785。
实施例6
本实施例中的β-酰基-烯丙基甲硫醚衍生物为2-((甲硫基)甲基)-1-(4-硝基苯基)丙-2-烯-1-酮。
其制备方法包括以下步骤:
1)将4-硝基苯乙酮(0.0413g,0.25mmol)、dmso(0.5ml)、无水醋酸钠(0.0164g,0.2mmol)、四氢吡咯烷(0.0209ml,0.25mmol)、对甲苯磺酸-水合物(0.0476g,0.25mmol)和无水醋酸(2ml)放入干净干燥的密闭反应容器中,在室温下混合均匀,随后在150℃条件下反应2小时;
2)反应完成后将反应物冷却至室温,用40ml乙酸乙酯将反应体系稀释溶解,加入20ml水,摇晃,静置,移除下面的水相后,再加入20ml饱和食盐水,摇晃,静置后,移除下面水相,有机相经无水硫酸钠干燥后,过滤,减压旋去溶剂;
3)将旋去溶剂后的反应物用硅胶柱层析(石油醚/乙酸乙酯=5:1,v/v)分离纯化,旋蒸除去溶剂,油泵抽干,既得黄色油状目标产物53.6g,产率85%。如图11和12所示,1hnmr(600mhz,cdcl3)δ8.28(d,j=8.5hz,2h),7.86(d,j=8.5hz,2h),6.02(s,1h),5.67(s,1h),3.51(s,2h),2.11(s,3h);13cnmr(151mhz,cdcl3)δ195.03,149.73,143.50,142.69,130.12,127.58,123.44,34.71,15.54.hrms(esi+):计算值c11h12no3s[m+h]+238.0532,实测值238.0537。
实施例7
本实施例中的β-酰基-烯丙基甲硫醚衍生物为1-([1,1'-联苯]-4-基)-2-((甲硫基)甲基)丙-2-烯-1-酮。
其制备方法包括以下步骤:
1)将联苯单乙酮(0.0491g,0.25mmol)、dmso(0.5ml)、无水醋酸钠(0.0164g,0.2mmol)、四氢吡咯烷(0.0209ml,0.25mmol)、对甲苯磺酸-水合物(0.0476g,0.25mmol)和无水醋酸(2ml)放入干净干燥的密闭反应容器中,在室温下混合均匀,随后在150℃条件下反应2小时;
2)反应完成后将反应物冷却至室温,用40ml乙酸乙酯将反应体系稀释溶解,加入20ml水,摇晃,静置,移除下面的水相后,再加入20ml饱和食盐水,摇晃,静置后,移除下面水相,有机相经无水硫酸钠干燥后,过滤,减压旋去溶剂;
3)将旋去溶剂后的反应物用硅胶柱层析(石油醚/乙酸乙酯=5:1,v/v)分离纯化,旋蒸除去溶剂,油泵抽干,既得黄色油状目标产物52.7g,产率79%。如图13和1所示,1hnmr(600mhz,cdcl3)δ7.88(d,j=8.2hz,2h),7.68(d,j=8.2hz,2h),7.63(d,j=7.6hz,2h),7.48(t,j=7.6hz,2h),7.40(t,j=7.3hz,1h),5.93(s,1h),5.72(s,1h),3.57(s,2h),2.15(s,3h);13cnmr(151mhz,cdcl3)δ196.48,145.27,143.81,139.85,136.05,130.12,128.89,128.12,127.20,126.92,125.43,35.37,15.49.hrms(esi+):计算值c17h17nos[m+h]+269.0995,实测值269.0991。
实施例8
本实施例中的β-酰基-烯丙基甲硫醚衍生物为1-(3-溴苯基)-2-((甲硫基)甲基)丙-2-烯-1-酮。
其制备方法包括以下步骤:
1)将3-溴苯乙酮(0.0331ml,0.25mmol)、dmso(0.5ml)、无水醋酸钠(0.0164g,0.2mmol)、四氢吡咯烷(0.0209ml,0.25mmol)、对甲苯磺酸-水合物(0.0476g,0.25mmol)和无水醋酸(2ml)放入干净干燥的密闭反应容器中,在室温下混合均匀,随后在150℃条件下反应2小时;
2)反应完成后将反应物冷却至室温,用40ml乙酸乙酯将反应体系稀释溶解,加入20ml水,摇晃,静置,移除下面的水相后,再加入20ml饱和食盐水,摇晃,静置后,移除下面水相,有机相经无水硫酸钠干燥后,过滤,减压旋去溶剂;
3)将旋去溶剂后的反应物用硅胶柱层析(石油醚/乙酸乙酯=5:1,v/v)分离纯化,旋蒸除去溶剂,油泵抽干,既得黄色油状目标产物49.7g,产率73%。如图15和16所示,1hnmr(600mhz,cdcl3)δ7.87(s,1h),7.67(d,j=7.8hz,2h),7.33(t,j=7.8hz,1h),5.95(s,1h),5.67(s,1h),3.51(s,2h),2.12(s,3h);13cnmr(151mhz,cdcl3)δ195.32,143.50,139.31,135.27,132.24,129.84,127.96,126.52,122.50,35.07,15.55.hrms(esi+):计算值c11h12bros[m+h]+270.9787,实测值270.9757。
实施例9
本实施例中的β-酰基-烯丙基甲硫醚衍生物为2-((甲硫基)甲基)-1-(吡啶-3-基)丙-2-烯-1-酮。
其制备方法包括以下步骤:
1)将3-乙酰吡啶(0.0275ml,0.25mmol)、dmso(0.5ml)、无水醋酸钠(0.0164g,0.2mmol)、四氢吡咯烷(0.0209ml,0.25mmol)、对甲苯磺酸-水合物(0.0476g,0.25mmol)和无水醋酸(2ml)放入干净干燥的密闭反应容器中,在室温下混合均匀,随后在150℃条件下反应2小时;
2)反应完成后将反应物冷却至室温,用40ml乙酸乙酯将反应体系稀释溶解,加入20ml水,摇晃,静置,移除下面的水相后,再加入20ml饱和食盐水,摇晃,静置后,移除下面水相,有机相经无水硫酸钠干燥后,过滤,减压旋去溶剂;
3)将旋去溶剂后的反应物用硅胶柱层析(石油醚/乙酸乙酯=1:1,v/v)分离纯化,旋蒸除去溶剂,油泵抽干,既得黄色油状目标产物26.5g,产率55%。如图17和18所示,1hnmr(600mhz,cdcl3)δ8.91(s,1h),8.73(d,j=4.8hz,1h),8.07–7.97(m,1h),7.38(dd,j=7.8,4.9hz,1h),5.97(s,1h),5.68(s,1h),3.49(s,2h),2.09(s,3h);13cnmr(151mhz,cdcl3)δ195.07,152.83,150.32,143.56,136.62,132.96,126.84,123.25,34.80,15.40.hrms(esi+):计算值c10h12nos[m+h]+194.0634,实测值194.0634。
实施例10
本实施例中的β-酰基-烯丙基甲硫醚衍生物为1-(5-溴噻吩-2-基)-2-((甲硫基)甲基)丙-2-烯-1-酮。
其制备方法包括以下步骤:
1)将2-乙酰基-5-溴噻吩(0.0513g,0.25mmol)、dmso(0.5ml)、无水醋酸钠(0.0164g,0.2mmol)、四氢吡咯烷(0.0209ml,0.25mmol)、对甲苯磺酸-水合物(0.0476g,0.25mmol)和无水醋酸(2ml)放入干净干燥的密闭反应容器中,在室温下混合均匀,随后在150℃条件下反应2小时;
2)反应完成后将反应物冷却至室温,用40ml乙酸乙酯将反应体系稀释溶解,加入20ml水,摇晃,静置,移除下面的水相后,再加入20ml饱和食盐水,摇晃,静置后,移除下面水相,有机相经无水硫酸钠干燥后,过滤,减压旋去溶剂;
3)将旋去溶剂后的反应物用硅胶柱层析(石油醚/乙酸乙酯=5:1,v/v)分离纯化,旋蒸除去溶剂,油泵抽干,既得黄色油状目标产物43.6g,产率63%。如图19和20所示,1hnmr(600mhz,cdcl3)δ7.43(s,1h),7.10(s,1h),5.81(s,1h),5.79(s,1h),3.47(s,2h),2.07(s,3h);13cnmr(151mhz,cdcl3)δ187.08,144.69,143.57,134.03,131.08,123.33,123.30,35.54,15.31.hrms(esi+):计算值c9h10bros2[m+h]+276.9351,实测值276.9361。
实施例11
本实施例中的β-酰基-烯丙基甲硫醚衍生物为1-(苯并呋喃-2-基)-2-((甲硫基)甲基)丙-2-烯-1-酮。
其制备方法包括以下步骤:
1)将2-乙酰基苯并呋喃(0.0400g,0.25mmol)、dmso(0.5ml)、无水醋酸钠(0.0164g,0.2mmol)、四氢吡咯烷(0.0209ml,0.25mmol)、对甲苯磺酸-水合物(0.0476g,0.25mmol)和无水醋酸(2ml)放入干净干燥的密闭反应容器中,在室温下混合均匀,随后在150℃条件下反应2小时;
2)反应完成后将反应物冷却至室温,用40ml乙酸乙酯将反应体系稀释溶解,加入20ml水,摇晃,静置,移除下面的水相后,再加入20ml饱和食盐水,摇晃,静置后,移除下面水相,有机相经无水硫酸钠干燥后,过滤,减压旋去溶剂;
3)将旋去溶剂后的反应物用硅胶柱层析(石油醚/乙酸乙酯=5:1,v/v)分离纯化,旋蒸除去溶剂,油泵抽干,既得黄色油状目标产物50g,产率86%。如图21和22所示,1hnmr(600mhz,cdcl3)δ7.71(d,j=7.8hz,1h),7.60(d,j=8.3hz,1h),7.49(dd,j=15.6,8.2hz,2h),7.32(t,j=7.5hz,1h),6.16(s,1h),5.94(s,1h),3.56(s,2h),2.11(s,3h);13cnmr(151mhz,cdcl3)δ184.58,155.98,151.96,143.67,128.39,126.78,125.03,123.93,123.27,115.86,112.49,35.23,15.36.hrms(esi+):计算值c13h13o2s[m+h]+233.0631,实测值233.0632。
上述的实施例中的无水醋酸钠还可以用醋酸锂、醋酸钾、醋酸铯或氟化钠中的任一种代替。
- 还没有人留言评论。精彩留言会获得点赞!