一种凝血因子X激活剂及其制备方法与流程
本发明属于生物医药领域,具体涉及一种来源于矛头蝮蛇(bothropsatrox)蛇毒的凝血因子ⅹ激活剂,本发明还涉及该激活剂的制备方法及其用途。
背景技术:
蛇毒中富含各种作用于哺乳动物凝血系统的诸多环节的蛋白酶和多肽,这些成分对凝血因子具有特异性,在抗凝、抗血栓和止血等方面有广泛的应用。凝血因子ⅹ激活剂(fⅹa)特异性作用于凝血因子ⅹ(fⅹ),它使fⅹ迅速转化为激活的凝血因子ⅹ(fⅹa),凝血酶原在fⅹa的作用下被激活形成具有活性的凝血酶,凝血酶作用于血浆纤维蛋白原使其转化为纤维蛋白,进而发挥止血的作用。从蛇毒中获得的凝血因子ⅹ激活剂比现有的止血药更有效,在临床手术止血具有巨大应用价值。fⅹa在蝰亚科、蝮亚科、眼镜蛇科等蛇毒中分布广泛。
从蛇毒中分离止血药具有悠久历史,目前临床应用较为成功的有瑞士生产的reptilase(中文商品名:立止血),它是从巴西矛头蝮蛇蛇毒中提取的类凝血酶和少量的凝血因子ⅹ激活剂的混合物。国内有立止血仿制品-巴曲亭,也是从矛头蝮蛇蛇毒中提取的类凝血酶制成的酶类止血药。国内也有将凝血因子ⅹ激活剂作为蝰蛇蛇毒试剂盒,用来诊断血浆中是否缺乏凝血因子ⅹ。
有关蛇毒凝血因子ⅹ激活剂的研究,国内外均有报道,研究最广泛和最深入的是蝰蛇蛇毒,如杨丽娟等以泰国产的圆斑蝰蛇蛇毒为原料,采用cm-sephadexc-50(弱阳离子交换填料)→sephadexg-75→superdex75三步纯化分离纯化得到一种fⅹ激活剂vrs-ⅹ,在还原和非还原条件下,经sds-page测定vrs-ⅹ为一条肽链,测定分子量为61.1±1.5kda,60.4±1.9kda(详见杨丽娟等,圆斑蝰蛇泰国亚种(viperarussellisiamensis)蛇毒凝血因子ⅹ激活剂的纯化及部分特性研究[j]福建医科大学学报,2002,36(3):242-246)。谢芸等以广西圆斑蝰蛇蛇毒为原料,依次经deaeff→superdeⅹg75两步层析分离纯化得到一种fⅹ激活剂(factorⅹactivatorofrussell’svipervenom,rvv-ⅹ),其在sds-page非还原性条件下呈单一蛋白条带,分子量约为97.8kda,还原性条件下呈二条蛋白条带,为一条重链(约76.3kda)和一条轻链(约21.5kda)(谢芸,许云禄,广西圆斑蝰蛇毒凝血因子ⅹ激活剂的分离纯化及其止血作用的研究[j]中国实用医药,2010,5(24):9-11)。1976年furukawa等依次用sephadexg-50→deaecellulose→sephadexg-200三步层析从蝰蛇蛇毒(russell’svipervenom)中获得rvv-ⅹ,其在sds-page还原性条件下呈四条蛋白条带,分别为61000、19800、16500和13000da(furukawa,y.,matsunaga,y.,hayashi,k.(1976).purificationandcharacterizationofacoagulantproteinfromthevenomofrussell’sviper.biochim.biophys.acta453:48-61)。kisiel等用sephadexg-150和qae-sephadexa-50两步层析从蝰蛇蛇毒(russell’svipervenom)中获得rvv-ⅹ,其在sds-page还原性条件下呈三条蛋白条带,分别为5900、20000和18000da(kisiel,w.,hermodson,m.a.,davie,e.w.(1976).factorⅹactivatingenzymefromrussell’svipervenom:isolationandcharacterization.biochemistry15(22):4901–4906)。
国内涉及从蝰蛇蛇毒中分离蛇毒凝血因子ⅹ激活剂(rvv-ⅹ)的相关专利,如发明名称为“蝰蛇蛇毒血凝酶的层析纯化方法”的中国发明专利cn02153855公开了rvv-ⅹ纯化过程为3步层析,依次用heparinsepharosecl-6b(肝素亲和柱),deae-sepharosefastflow(阴离子交换)和sephacryl-300hr(凝胶过滤)填料。但是,该方法中,由于蛇毒原料成分复杂,第一步用亲和柱对填料的损伤较大,而第三步用sephacryl-300hr(凝胶过滤)填料,此填料属软胶填料,耐压性较弱,层析过程中流速较慢,无法进一步放大生产。
发明名称为“一种止血药”的中国发明专利03101881.5公开了对rvv-ⅹ纯化的技术方案,其是通过使用sephacryl系列凝胶过滤柱粗分和q-sepharose系列离子交换柱两个步骤实现的,虽然该申请中并没有描述蛇毒来源,但是根据其公开的蛇毒凝血因子ⅹ激活剂(rvv-ⅹ),可以推测蛇毒来源是蝰蛇类。
发明名称为“一种高效提取凝血因子十活化酶的方法”的中国发明专利申请98119978.x公开了rvv-ⅹ纯化的技术方案,其通过monoq柱(阴离子交换柱)一步层析从蝰蛇毒中获得电泳纯样品。
发明名称为“圆斑蝰蛇蛇毒凝血ⅹ因子激活酶及其提取方法与应用”的中国发明专利申请200710102028.6公开了对rvv-ⅹ纯化的技术方案,依次用deae-sephadexa-50阴离子交换层析、纤维素deae-32阴离子交换层析和superose-12凝胶过滤层析,从圆斑蝰蛇毒中制备得到。该方法中所使用的纤维素deae-32阴离子交换填料为软胶,其柱体积会随缓冲液中离子强度变化而变化,因此,该方法的层析过程可控性差,不利于规模化生产。
发明名称为“一种具有促凝血作用的复方生物组分及其药物制剂”的中国发明专利200410020115.3公开了凝血酶和类凝血激酶(凝血因子十活化酶)分离纯化的方法,纯化过程为sephadexg-25(凝胶过滤柱)层析后,再进行qae-sephadexa50(阴离子交换柱)层析,第一步层析用分子筛凝胶填料,该方法的缺点是严重限制了蛇毒的上样量,且层析过程耗时较长,无法进一步放大生产。
而关于矛头蝮蛇来源的凝血因子ⅹ激活剂的研究相对较少,hofmann等采用deaecellulose→冻干透析→sephadexg-100步层析获得fⅹ激活剂(hofmann,h.,dumarey,c.,bon,c.(1983).bloodcoagulationinducedbybothropsatroxvenom.identificationandpropertiesofafactorⅹactivator.biochimie65(3):201–210)。发明名称为“一种原于蛇毒的复方止血药”中国发明专利申请cn200510047332中分离纯化fⅹa的蛇毒来源是矛头蝮蛇蛇毒,用deae-fastflow(阴离子交换填料)和sephacryl系列(凝胶过滤填料)填料纯化实现。显然地,当前缺乏从矛头蝮蛇获得凝血因子ⅹ激活剂的研究。
综上所述,当前的凝血因子ⅹ激活剂的纯化原料主要是从蝰蛇毒中分离纯化,并且其制备方法繁琐,极大的增加了生产成本,且无法用于规模化的工业生产。
因此,当前对新的凝血因子ⅹ激活剂以及更可控的制备方法存在需求。
技术实现要素:
因此,本发明的目的是针对现有技术的不足,提供一种凝血因子ⅹ激活剂,本发明还提供了该凝血因子ⅹ激活剂的制备方法。本发明提供的凝血因子ⅹ激活剂纯度高,使用体积排阻色谱sec柱检测,纯度为100%,c4反相柱检测,纯度高达96%,可以广泛用于制备治疗外科出血、内科出血、妇产科出血、五官科出血、儿科出血和组织活检后出血等临床出血及出血性疾病的药物,将在生物制药领域发挥重要作用,应用前景广阔,本发明提供的凝血因子ⅹ激活剂的制备方法工艺可控,可以进行规模化生产。
一方面,本发明提供了一种凝血因子ⅹ激活剂,所述凝血因子ⅹ激活剂来源于矛头蝮蛇(bothropsatrox)蛇毒,其中,所述凝血因子ⅹ激活剂的等电点为4.94-6.64;在非还原sds-page中,所述凝血因子ⅹ激活剂呈单一条带,测定分子量为70-96kda,更优选地为75-88kda;在还原sds-page中,所述凝血因子ⅹ激活剂呈三条带,分别为h链、l1链和l2链,测定分子量分别为h链:50-64kda;l1链:11-16kda;l2链:11-16kda;
优选地,所述凝血因子ⅹ激活剂的h链n-末端序列如seqidno:1所示:seqidno:1ltpeqqayldakkyv;
优选地,所述凝血因子ⅹ激活剂的l1链n-末端如seqidno:2所示:
seqidno:2dcpsdwssyeghcyr;
优选地,所述凝血因子ⅹ激活剂的l2链n-末端序列如seqidno:3所示:
seqidno:3dcnsywstyegrsyr;
优选地,所述凝血因子ⅹ激活剂的糖酶切显示糖位点在重链,为n-糖;优选地,maldi-tof测定n-糖基化含量范围为25-34%,更优选地为27-33%,进一步优选地为28.8%-31.7%;
优选地,maldi-tof测定所述凝血因子ⅹ激活剂的分子量为85-90kda,更优选地为86-88kda,进一步优选地为87.4kda-87.6kda;
优选地,esi-q-tof测定所述凝血因子ⅹ激活剂的分子量为84-90kda;更优选地为86-89kda,进一步优选地为85.7kda-88.2kda;
优选地,maldi-tof测定所述凝血因子ⅹ激活剂的脱糖分子量为59-62kda,更优选地为61kda;
优选地,esi-q-tof测定l1链的分子量为13-16kda,更优选地为14-15kda,进一步优选地为14.7-14.8kda;
优选地,esi-q-tof测定l2链的分子量为13-16kda,更优选地为14-15kda,进一步优选地为14.5kda;
优选地,esi-q-tof测定h链的分子量为52-64kda,更优选地为55-62kda。
另一方面,本发明提供了一种凝血因子ⅹ激活剂的制备方法,所述方法包括以下步骤:
将矛头蝮蛇蛇毒溶解,离心后取上清液,依次进行阴离子交换层析、阳离子交换层析、亲和或复合填料层析、阴离子交换层析、凝胶过滤层析,得到凝血因子ⅹ激活剂。
优选地,本发明所述的方法包括以下步骤:
1)将矛头蝮蛇蛇毒溶于ph7.1-9.0、浓度为0.01-0.1mol/l的碱性缓冲液,得到浓度为20-150mg/ml的溶液,离心后取上清液;
2)将步骤1)得到的上清液进行阴离子交换层析,线性梯度洗脱,收集含有凝血因子ⅹ激活剂活性洗脱峰的洗脱液,使用截留分子量为5000~30000kda的膜包和透析液对洗脱液进行透析;
其中,所述线性梯度洗脱中,梯度洗脱剂a为ph为7.0-8.9,浓度为0.01-0.1mol/l的碱性缓冲液;梯度洗脱剂b为含有0.1-0.6mol/lnacl的、ph为7.0-8.9、浓度为0.01-0.1mol/l的碱性缓冲液;
其中,所述透析液为ph为5.0-6.8,浓度为0.02-0.1mol/l的酸性缓冲液;
优选地,所述阴离子交换层析的填料选自填料配基为季胺、叔胺、仲胺和伯胺基团类型的填料;更优选地,所述阴离子交换层析填料为deae-sepharosefastflow;
优选地,将透析后的洗脱液保存在4℃;
3)将步骤2)透析后的洗脱液进行阳离子交换层析,线性梯度洗脱,收集含有凝血因子ⅹ激活剂活性洗脱峰的洗脱液,使用截留分子量为5000~30000kda的膜包和透析液对洗脱液进行透析;
其中,所述线性梯度洗脱中,梯度洗脱剂a为ph为5.0-6.8,浓度为0.02-0.1mol/l的酸性缓冲液,梯度洗脱剂b为含有0.1-0.6mol/lnacl的、ph为5.0-6.8、浓度为0.02-0.1mol/l的酸性缓冲液;
优选地,对应于下一步的亲和层析或复合填料层析,所述透析液选自亲和层析透析液或复合填料层析透析液;更优选地,所述亲和层析透析液为含0-2mol/lnacl、0.001-0.005mol/lcacl2和mncl2的、ph为7.0-8.5、浓度为0.01-0.1mol/l的碱性缓冲液;所述复合填料层析透析液为含0-0.5mol/l氯化钠的、ph为7.0-8.5、浓度为0.01-0.1mol/l的碱性缓冲液;
优选地,所述阳离子交换层析的填料选自填料配基为磺酸基团类、磷酸或亚磷酸基团类、酚羟基和羧基类的填料;更优选地,所述阳离子填料为sp-sepharosefastflow;
优选地,将透析后的洗脱液保存在4℃;
4)将步骤3)透析后的洗脱液进行亲和层析或复合填料层析,线性梯度洗脱,收集含有凝血因子ⅹ激活剂活性洗脱峰的洗脱液,使用截留分子量为5000~30000kda的膜包和透析液对洗脱液进行透析;
其中,所述亲和层析的固相填充物选自含有两类配基的亲和填料;所述复合填料层析的复合填料选自羟基磷灰石类;
其中,所述亲和层析的线性梯度洗脱中,梯度洗脱剂a为含0-2mol/lnacl、ph为7.0-8.5、浓度为0.01-0.1mol/l的碱性缓冲液;梯度洗脱剂b为含0-2mol/l氯化钠和0.05-0.5mol/l糖类竞争性洗脱剂、ph7.0-8.5、浓度为0.01-0.1mol/l碱性缓冲液;
其中复合填料层析的线性梯度洗脱中,梯度洗脱剂a为含有0-0.5mol/lnacl的、ph7.0-8.5、浓度为0.01-0.1mol/l的碱性缓冲液,梯度洗脱剂b为含0.6-2mol/lnacl、ph7.0-8.5、浓度为0.01-0.1mol/l的碱性缓冲液。
其中,所述透析液为含有0-0.1mol/lnacl的、ph7.0-8.9、浓度为0.01-0.1mol/l的碱性缓冲液,
优选地,将透析后的洗脱液保存在4℃;
5)将步骤4)透析后的洗脱液进行阴离子交换层析,线性梯度洗脱,收集含有凝血因子ⅹ激活剂活性洗脱峰的洗脱液,使用截留分子量为5000~30000kda的膜包和透析液对洗脱液进行透析;
其中,所述线性梯度洗脱中,梯度洗脱剂a为含有0-0.1mol/lnacl的、ph为7.0-8.9,浓度为0.01-0.1mol/l的碱性缓冲液;梯度洗脱剂b为含有0.1-0.6mol/lnacl的、ph为7.0-8.9、浓度为0.01-0.1mol/l的碱性缓冲液;
其中,所述透析液为含有0-0.1mol/lnacl的、ph为7.0-8.5,浓度为0.01-0.1mol/l的碱性缓冲液;
优选地,所述阴离子交换层析填料选自填料配基为季铵、叔胺、仲胺和伯胺基团类型的填料;更优选地,所述阴离子交换层析填料选自deae-sepharosefastflow或captodeae;
优选地,将透析后的洗脱液保存在4℃;
6)将透析后的洗脱液进行凝胶过滤层析,直线洗脱,收集含有凝血因子ⅹ激活剂活性洗脱峰的洗脱液,得到凝血因子ⅹ激活剂;
优选地,所述凝胶为superdexg75;
其中,所述洗脱剂为含有0-0.1mol/lnacl的,ph为7.0-8.5、浓度为0.01-0.1mol/l的碱性缓冲液。
优选地,所述收集含有凝血因子ⅹ激活剂活性洗脱峰的洗脱液通过包括以下步骤的方法实现的:
收集洗脱液,在280nm的波长下对洗脱液进行紫外分光光度计检测,依紫外吸收图谱收集各洗脱峰并测定活性,sds-page凝胶电泳跟踪检测,收集含有凝血因子ⅹ激活剂活性洗脱峰的洗脱液;更优选地,所述活性测定中使用s-2765底物生色法(参照专利申请号cn201510940717.9);
优选地,在步骤1)中,所述碱性缓冲液的ph为7.5-8.5、浓度为0.04-0.06mol/l;进一步优选地,所述碱性缓冲液的ph为8.0、浓度为0.05mol/l;
优选地,在步骤1)中,所述离心的条件为:4-8℃下,相对离心力为1200g-20000g,优选地为3000g-4000g,离心时间为5-20min,优选地为15min;
优选地,在步骤2)中,所述梯度洗脱剂a为ph为7.4-8.5、浓度为0.04-0.06mol/l的碱性缓冲液;更优选地,所述梯度洗脱剂a为ph为8.0、浓度为0.05mol/l的碱性缓冲液;
所述梯度洗脱剂b为含有0.4-0.6mol/lnacl的、ph为7.4-8.5、浓度为0.04-0.06mol/l的碱性缓冲液;更优选地,所述梯度洗脱剂b为含有0.5mol/lnacl的、ph为8.0、浓度为0.05mol/l的碱性缓冲液;
优选地,在步骤2)中,在进行阴离子交换层析之前,使用ph为7.0-8.9,浓度为0.01-0.1mol/l的碱性缓冲液作为平衡液平衡10个柱体积及以上;优选地,所述平衡液为ph为7.4-8.5、浓度为0.04-0.06mol/l的碱性缓冲液;更优选地,所述平衡液为ph为8.0、浓度为0.05mol/l的碱性缓冲液;
优选地,在步骤2)中,平衡时线性流速为60-200cm/h,更优选地为100cm/h;
优选地,在步骤2)中,洗脱时,线性流速为20-100cm/h,更优选地为60cm/h;
优选地,在步骤2)中,所述透析液为ph为5.5-6.5、浓度为0.04-0.08mol/l的酸性缓冲液;进一步优选地,所述透析液为ph为6.0、浓度为0.05mol/l的酸性缓冲液;
优选地,在步骤3)中,所述梯度洗脱剂a为ph为5.5-6.5、浓度为0.04-0.08mol/l的酸性缓冲液;进一步优选地,所述梯度洗脱剂a为ph为6.0、浓度为0.05mol/l的酸性缓冲液;
更优选地,所述梯度洗脱剂b为含有0.4-0.6mol/lnacl的、ph为5.5-6.5的、浓度为0.04-0.08mol/l的酸性缓冲液;进一步优选地,所述梯度洗脱剂b为含有0.5mol/lnacl的、ph为6.0、浓度为0.05mol/l的酸性缓冲液;
优选地,在步骤3)中,在进行阳离子交换层析之前,使用ph为5.0-6.8,浓度为0.02-0.1mol/l的酸性缓冲液作为平衡液平衡10个柱体积及以上;优选地,所述平衡液为ph为5.5-6.5、浓度为0.04-0.08mol/l的酸性缓冲液;进一步优选地,所述平衡液为ph为6.0、浓度为0.05mol/l的酸性缓冲液;
优选地,在步骤3)中,平衡时线性流速为60-200cm/h,更优选地为100cm/h;
优选地,在步骤3)中,洗脱时,线性流速为20-100cm/h,更优选地为60cm/h;
优选地,在步骤3)中,所述亲和层析透析液为含有0.2-1mol/lnacl、0.001-0.003mol/lcacl2和mncl2的、ph为7.6-8.4、浓度为0.02-0.05mol/l的碱性缓冲液;进一步优选地,所述透析液为含有0.8molnacl/l、0.002mol/lcacl2和mncl2的、ph为8.0、浓度为0.03mol/l的碱性缓冲液;
优选地,在步骤3)中,所述复合填料层析透析液为含有0.2-0.4mol/l氯化钠的、ph为7.6-8.4、浓度为0.02-0.05mol/l的碱性缓冲液;进一步优选地,所述透析液为含有0.3mol/l氯化钠、ph为8.0、浓度为0.03mol/l的碱性缓冲液;
优选地,在步骤4)中,所述亲和层填料配基分别为扁豆外源凝集素类和刀豆球蛋白类;更优选地为lentillectinsepharose4b、conasepharose4b或6b、进一步优选地为lentillectinsepharose4b和conasepharose4b。
优选地,在步骤4)中,所述羟基磷灰石复合类填料选自羟基氟化磷灰石、羟基磷灰石和氟代磷灰石填料;更优选地为cht陶瓷羟基磷灰石i;
优选地,在步骤4)中,所述亲和层析中,梯度洗脱剂a为含有0.2-1mol/lnacl、ph为7.6-8.4、浓度为0.02-0.05mol/l的碱性缓冲液;进一步优选地,所述梯度洗脱剂a为含有0.8molnacl/l、ph为8.0、浓度为0.03mol/l的碱性缓冲液;所述亲和层析中,梯度洗脱剂b为含有0.1-1mol/lnacl和0.1-0.5mol/l糖类竞争性洗脱剂的、ph为7.6-8.4、浓度为0.02-0.05mol/l的碱性缓冲液;更优选地,所述梯度洗脱剂b为含有0.8mol/lnacl和0.2-0.4mol/l糖类竞争性洗脱剂的、ph为8.0、浓度为0.03mol/l的碱性缓冲液;
优选地,在步骤4)中,所述复合填料层析中,梯度洗脱剂a为含有0.2-0.4mol/lnacl的、ph为7.6-8.4、浓度为0.02-0.05mol/l的碱性缓冲液;更优选地,所述梯度洗脱剂a为含有0.3mol/lnacl的、ph为8.0、浓度为0.03mol/l的碱性缓冲液;
所述复合填料层析中,梯度洗脱剂b为含有0.6-1mol/lnacl的、ph为7.6-8.4、浓度为0.02-0.05mol/l的碱性缓冲液;更优选地,所述梯度洗脱剂b为含有0.8mol/lnacl、ph为8.0、浓度为0.03mol/l的碱性缓冲液;
优选地,在步骤4)中,所述糖类竞争性洗脱剂选自甲基葡萄糖类竞争性洗脱剂和甲基吡喃糖类竞争性洗脱剂;
优选地,在步骤4)中,在进行亲和层析之前,使用含0-2mol/lnacl的、0.001-0.005mol/lcacl2和mncl2的、ph为7.0-8.5、浓度为0.01-0.1mol/l的碱性缓冲液作为平衡液平衡10个柱体积及以上;优选地,所述平衡液为含有0.2-1mol/lnacl、0.001-0.003mol/lcacl2和mncl2的、ph为7.6-8.4、浓度为0.02-0.05mol/l的碱性缓冲液;进一步优选地,所述平衡液为0.8mol/lnacl、0.002mol/lcacl2和mncl2的、ph为8.0、浓度为0.03mol/l的碱性缓冲液;
优选地,在步骤4)中,在进行复合填料层析之前,使用含有0-0.5mol/lnacl的、ph7.0-8.5、浓度为0.01-0.1mol/l的碱性缓冲液作为平衡液平衡10个柱体积及以上;优选地,所述平衡液为含0.2-0.4mol/lnacl的、ph为7.6-8.4、浓度为0.02-0.05mol/l的碱性缓冲液;进一步优选地,所述平衡液为含0.3mol/lnacl的ph为8.0、浓度为0.03mol/l的碱性缓冲液;
优选地,在步骤4)中,亲和层析平衡时线性流速为30-70cm/h,更优选地为60cm/h;
优选地,在步骤4)中,亲和层析洗脱时线性流速为30-70cm/h,更优选地为40cm/h;
优选地,在步骤4)中,复合填料层析平衡时线性流速为50-200cm/h,更优选地为100cm/h;
优选地,在步骤4)中,复合层析洗脱时线性流速为50-100cm/h,更优选地为80cm/h;
优选地,在步骤4)中,所述透析液为含有0.05-0.1mol/lnacl的、ph为7.5-8.5、浓度为0.02-0.05mol/l的碱性缓冲液;进一步优选地,所述透析液为含有0.1mol/l的nacl的、ph为8.0、浓度为0.03mol/l的碱性缓冲液;
优选地,在步骤5)中,所述梯度洗脱剂a为含有0.05-0.1mol/l的nacl的、ph为7.5-8.5、浓度为0.02-0.05mol/l的碱性缓冲液;更优选地,所述梯度洗脱剂a为含有0.1mol的nacl的、ph为8.0、浓度为0.03mol/l的碱性缓冲液;
所述梯度洗脱剂b为含有0.4-0.6mol/lnacl的、ph为7.5-8.5、浓度为0.02-0.05mol/l的碱性缓冲液;更优选地,所述梯度洗脱剂b为含有0.5mol/lnacl的、ph为8.0、浓度为0.03mol/l的碱性缓冲液;
优选地,在步骤5)中,在进行阴离子交换层析之前,使用含有0-0.1mol/lnacl的、ph为7.0-8.9,浓度为0.01-0.1mol/l的碱性缓冲液作为平衡液平衡10个柱体积及以上;优选地,所述平衡液为ph为含有0.05-0.1mol/l的nacl的、7.5-8.5、浓度为0.02-0.05mol/l的碱性缓冲液;更优选地,所述平衡液为含有0.1mol/l的nacl的、ph为8.0、浓度为0.03mol/l的碱性缓冲液;
优选地,在步骤5)中,平衡时线性流速为60-200cm/h,更优选地为100cm/h;
优选地,在步骤5)中,洗脱时,线性流速为20-100cm/h,更优选地为60cm/h;
优选地,在步骤5)中,所述透析液为含有0.01-0.05mol/l的nacl的、ph为7.5-8.5、浓度为0.01-0.05mol/l的碱性缓冲液;进一步优选地,所述透析液为含有0.05mol/l的nacl的、ph为8.0、浓度为0.02mol/l的碱性缓冲液;
优选地,在步骤6)中,所述洗脱剂为含有0.01-0.05mol/l的nacl的、ph为7.5-8.5、浓度为0.01-0.05mol/l的碱性缓冲液;进一步优选地,所述洗脱剂为含有0.05mol/l的nacl的、ph为8.0、浓度为0.02mol/l的碱性缓冲液;
优选地,在步骤6)中,在进行凝胶过滤层析之前,使用含有0-0.1mol/lnacl的、ph为7.0-8.5,浓度为0.01-0.1mol/l的碱性缓冲液作为平衡液平衡5个柱体积及以上;优选地,所述平衡液为含有0.01-0.05mol/l的nacl的、ph7.5-8.5、浓度为0.01-0.05mol/l的碱性缓冲液;更优选地,所述平衡液为含有0.05mol/l的nacl的、ph为8.0、浓度为0.02mol/l的碱性缓冲液;
优选地,在步骤6)中,平衡时线性流速为20-80cm/h,更优选地为60cm/h;
优选地,在步骤6)中,洗脱时,线性流速为20-60cm/h,更优选地为40cm/h;
优选地,在步骤6)中,所述凝胶填料的分离范围为3000-300000da。
优选地,所述碱性缓冲液选自tris-hcl缓冲液、磷酸盐缓冲液和磷酸盐-柠檬酸缓冲液;
优选地,所述酸性缓冲液选自磷酸盐缓冲液、磷酸盐-柠檬酸缓冲液和醋酸盐缓冲液;
另一方面,本发明提供了本发明的凝血因子x激活剂在制备用于治疗出血病症的药物中的应用;
优选地,所述出血包括临床出血、手术出血和各种内外科急性出血。
再另一方面,本发明提供了一种用于治疗出血的药物组合物,所述药物组合物包括所述凝血因子x激活剂以及药学上可接受的载体。
与现有技术相比,本发明具备以下优点:
1)本发明提供的凝血因子x激活剂纯度高,使用体积排阻色谱sec柱检测,纯度为100%,使用c4反相柱检测,纯度高达96%。
2)本发明提供的方法首先使用阴阳离子交换填料层析,然后进行亲和层析,不仅可以使亲和柱层析后凝血因子x激活剂的纯度接近电泳纯,还极大地延长亲和填料的使用寿命,从而节约了生产成本,有利于大规模的工业化生产。
本领域技术人员已知的是,当前的大多数亲和填料的配基很容易在样品成分复杂的层析过程中脱落,并且这些填料的价格高,所以在实际应用中,亲和层析通常在蛋白粗分之后的精分或精细化分离步骤中使用。本发明的发明人发现,在制备凝血因子ⅹ激活剂时,前两步采用阴阳离子交换填料,可以除去大部分无凝血因子x激活剂活性组分,为下一步亲和层析提供较纯的上样样品,从而极大地减少了亲和层析过程中杂质蛋白的非特异性吸附,并提高了填料对凝血因子x激活剂的载量,也使亲和柱层析后凝血因子x激活剂的纯度接近电泳纯。
3)本发明的发明人发现特定的亲和填料能够与矛头蝮蛇蛇毒中的凝血因子x激活剂专一结合,从而极大提高了生产效率。
4)本发明提供的凝血因子ⅹ激活剂的制备方法特别适合用于生产人用药方式,本发明的方法所使用的层析柱可依据制备要求同比放大,工艺稳定,层析过程可控;
5)本发明的方法在透析过程中采用膜包或膜堆装置,透析时间短(2h内完成),方便,透析过程可保持无菌,并在透析过程中可以除去蛇毒溶液中如多肽等小分子杂质。
附图说明
以下,结合附图来详细说明本发明的实施方案,其中:
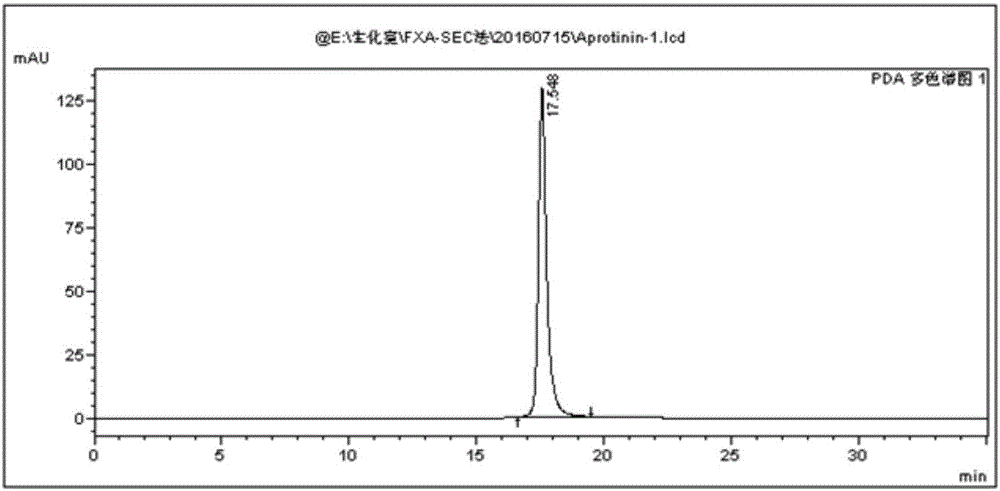
图1为本发明的凝血因子ⅹ激活剂的高效液相sec柱纯度检测图;
图2为本发明的凝血因子ⅹ激活剂的高效液相反相c4柱纯度检测;
图3为本发明的凝血因子ⅹ激活剂sds-page纯度(实施例一);
图4为本发明的凝血因子ⅹ激活剂sds-page纯度(实施例二);
图5为本发明的凝血因子ⅹ激活剂的maldi-tof分子量测定结果;
图6为本发明的凝血因子ⅹ激活剂的maldi-tof分子量质谱图
具体实施方式
下面的实施例将对本发明作进一步的解释说明,但是本发明并不仅仅局限于这些实施例,这些实施例仅作为更详细具体地说明之用,而不应理解为用于以任何形式限制本发明。本领域的技术人员在权力要求的范围内所作出的某些改变和调整也应认为属于本发明的范围。
实施例1矛头蝮蛇蛇毒凝血因子ⅹ激活剂的分离纯化
1、蛇毒溶解
取10g矛头蝮蛇蛇毒(蛇毒购自辽宁远大诺康生物制药有限公司),用100mlph8.0、0.05mol/l的tris-hcl缓冲液于4-8℃的层析柜中溶解后,3000g离心15min,取上清液备用。
2、deae-sepharosefastflow柱层析:2.6×20cm(填料购自ge公司)
2-1)deae-sepharosefastflow装柱:取保存在20%乙醇中的deae-sepharosefastflow填料约90ml,先用纯净水置换除醇后装填2.6×20cm层析柱,然后用ph8.0、0.05mol/l的tris-hcl缓冲液平衡10个柱体积以上,线性流速100cm/h,280nm波长紫外吸收检测。
2-2)上样、洗脱:将步骤2-1)中离心后的蛇毒上清液泵入已平衡好的层析柱,线性流速60cm/h,上样结束后,用ph8.0、0.05mol/l的tris-hcl缓冲液进行平衡洗脱,待紫外吸收降至基线后,进行线性梯度洗脱,梯度洗脱剂a为ph8.0、0.05mol/l的tris-hcl缓冲液300ml,梯度洗脱剂b为含0.5mol/l氯化钠的ph8.0、0.05mol/l的tris-hcl缓冲液300ml,线性流速60cm/h,依据280nm波长紫外检测图谱收集各峰,共收集8个层析峰。活性测定和凝胶电泳跟踪确定第6峰为ⅹ因子激活剂活性主峰。
2-3)样品透析:步骤2-2)收集的活性峰用截流分子量10000da的膜包(购自赛多利斯,聚醚砜材质)对ph6.0、0.05mol/l的磷酸盐缓冲液进行透析,样品定容至约100ml。置4℃冰箱中备用。
3、sp-sepharosefastflow柱层析:2.6×20cm(填料购自ge公司)
3-1)sp-sepharosefastflow装柱:取保存在20%乙醇中的sp-sepharosefastflow填料约90ml,先用纯净水置换除醇后装填2.6×20cm层析柱,然后用ph6.0、0.05mol/l的磷酸盐缓冲液平衡10个柱体积以上,线性流速100cm/h,280nm波长紫外吸收检测。
3-2)上样、洗脱:将步骤2中透析后的蛇毒溶液泵入已平衡好的层析柱,线性流速60cm/h,上样结束后,用ph6.0、0.05mol/l的磷酸盐缓冲液进行平衡洗脱,待紫外吸收降至基线后,进行线性梯度洗脱,梯度洗脱剂a为ph6.0、0.05mol/l的磷酸盐缓冲液300ml,梯度洗脱剂b为含0.5mol/l氯化钠的ph6.0、0.05mol/l的磷酸盐缓冲液300ml,线性流速60cm/h,依据280nm波长紫外检测图谱收集各峰,共收集4个层析峰。活性测定和凝胶电泳跟踪确定第3峰为x因子激活剂活性主峰。
3-3)样品透析:步骤3-2)收集的活性峰用截流分子量10000da的膜包对含0.8mol/l氯化钠、0.002mol/l氯化钙和0.002mol/l氯化锰的ph8.0、0.03mol/l的tris-hcl缓冲液进行透析,样品定容至约80ml。置4℃冰箱中备用。
4、lentillectinsepharose4b柱层析:1.6×20cm(填料购自ge公司)
4-1)lentillectinsepharose4b装柱:取保存在20%乙醇中的lentillectinsepharose4b填料约40ml,先用纯净水置换除醇后装填1.6×20cm层析柱,然后用含0.8mol/l氯化钠、0.002mol/l氯化钙和0.002mol/l氯化锰的ph8.0、0.03mol/l的tris-hcl缓冲液平衡10个柱体积以上,线性流速60cm/h,280nm波长紫外吸收检测。
4-2)上样、洗脱:将步骤3中透析后的蛇毒溶液泵入已平衡好的层析柱,线性流速40cm/h,上样结束后,用含0.8mol/l氯化钠、0.002mol/l氯化钙和0.002mol/l氯化锰的ph8.0、0.03mol/l的tris-hcl缓冲液平衡洗脱,待紫外吸收降至基线后,进行线性梯度洗脱,梯度洗脱剂a为含0.8mol/l氯化钠的ph8.0、0.03mol/l的tris-hcl缓冲液150ml,梯度洗脱剂b为含0.8mol/l氯化钠和0.4mol/l甲基-α-d-吡喃葡萄糖苷的ph8.0、0.03mol/l的tris-hcl缓冲液150ml,线性流速40cm/h,依据280nm波长紫外检测图谱收集各峰,共收集3个层析峰。活性测定和凝胶电泳跟踪确定第2峰为x因子激活剂活性主峰。
4-3)样品透析:步骤4-2)收集的活性峰用截流分子量10000da的膜包对含0.1mol/l氯化钠ph8.0、0.03mol/l的tris-hcl缓冲液进行透析,样品定容至约100ml。置4℃冰箱中备用。
5、deae-sepharosefastflow柱层析:1.6×20cm(填料购自ge公司)
5-1)deae-sepharosefastflow装柱:取保存在20%乙醇中的deae-sepharosefastflow填料约40ml,先用纯净水置换除醇后装填1.6×20cm层析柱,然后用含0.1mol/l氯化钠ph8.0、0.03mol/l的tris-hcl缓冲液平衡10个柱体积以上,线性流速100cm/h,280nm波长紫外吸收检测。
5-2)上样、洗脱:将步骤4中透析后的蛇毒溶液泵入已平衡好的层析柱,线性流速60cm/h小时,上样结束后,用含0.1mol/l氯化钠ph8.0、0.03mol/l的tris-hcl缓冲液进行平衡洗脱,待紫外吸收将至基线后,进行线性梯度洗脱,梯度洗脱剂a为含0.1mol/l氯化钠ph8.0、0.03mol/l的tris-hcl缓冲液150ml,梯度洗脱剂b为含0.5mol/l氯化钠的ph8.0、0.03mol/l的tris-hcl缓冲液150ml,线性流速60cm/h,依据280nm波长紫外检测图谱收集各峰,共收集3个层析峰。活性测定和凝胶电泳跟踪确定第2峰为ⅹ因子激活剂活性主峰。
5-3)样品透析:步骤5-2)收集的活性峰用截流分子量10000da的膜包对含0.05mol/l氯化钠ph8.0、0.02mol/l的tris-hcl缓冲液进行透析浓缩,样品定容至约50ml。置4℃冰箱中备用。
6、superdexg75柱层析:2.6×70cm(填料购自ge公司)
6-1)superdexg75装柱:取保存在20%乙醇中的superdexg75填料约350ml,先用纯净水置换除醇后装填2.6×70cm层析柱,然后用含0.05mol/l氯化钠的ph8.0、0.02mol/l的tris-hcl缓冲液平衡5个柱体积以上,线性流速60cm/h小时,280nm波长紫外吸收检测。
6-2)上样、洗脱:将步骤5中透析浓缩蛇毒溶液,用注射器吸取10ml样品液上样,沿柱侧壁旋转缓慢加入,待样品液完全进入填料后,用少于0.2ml的含0.05mol/l氯化钠的ph8.0、0.02mol/l的tris-hcl缓冲液润洗侧壁和胶面后,加入10ml缓冲液后进行洗脱,线性流速40cm/h。依据280nm下紫外分光广度检测图谱收集各峰,共收集1个层析峰。
7、结果分析:
7-1)纯度检测
采用高效液相色谱(岛津高效液相色谱仪20a,sec-hplc:g2000wxl;rp-hplc:c4色谱柱)和电泳系统进行检测,sec-hplc检测呈单一峰(如图1所示),rp-hplc检测呈单一峰(如图2所示),按照面积归一化法计算,凝血因子ⅹ激活剂含量分别为为100%和96.2%。sds-page变性非还原电泳检测目标蛋白组分显示单一条带(如图3所示)。
7-2)收率及比活
目标蛋白活性检测经s2765底物法检测,10g溶解后溶液中凝血因子ⅹ激活剂总活性为60万单位,经层析纯化后收集的活性为12万单位,收率为20%。比活为4491u/mg。
7-3)矛头蝮蛇凝血因子ⅹ激活剂的结构特征
a.maldi-tof质谱测得凝血因子ⅹ激活剂分子量为87540da,如图5和图6所示。
b.固相ph梯度(ipg)等电聚焦法(bio-rad手册)测得等电点为4.94-6.64;
c.还原sds-page和edman测序(测序仪:abiprocise491):
还原sds-page呈三条带,测定分子量分别为h:54kda;l1:12kda;l2:13kda。
h链n-末端序列:seqidno:1:ltpeqqayldakkyv;
l1链n-末端序列:seqidno:2:dcpsdwssyeghcyr;
l2链n-末端序列:seqidno:3:dcnsywstyegrsyr。
d.糖酶切显示糖位点在重链,为n-糖。
实施例2矛头蝮蛇蛇毒凝血因子ⅹ激活剂的分离纯化
1、蛇毒溶解
取10g矛头蝮蛇蛇毒(购自辽宁远大诺康生物制药有限公司),用100mlph7.4、0.01mol/l的tris-hcl缓冲液于4-8℃的层析柜中溶解后,3000g离心15min,取上清液备用。
2、deae-sepharosefastflow柱层析:2.6×20cm(购自ge公司)
2-1)deae-sepharosefastflow装柱:取保存在20%乙醇中的deae-sepharosefastflow填料约90ml,先用纯净水置换除醇后装填2.6×20cm层析柱,然后用ph7.4、0.01mol/l的tris-hcl缓冲液平衡10个柱体积以上,线性流速100cm/h,280nm波长紫外吸收检测。
2-2)上样、洗脱:将离心后的蛇毒上清液泵入已平衡好的层析柱,线性流速60cm/h,上样结束后,用ph7.4、0.01mol/l的tris-hcl缓冲液进行平衡洗脱,待紫外吸收降至基线后,进行线性梯度洗脱,梯度洗脱剂a为ph7.4、0.01mol/l的tris-hcl缓冲液300ml,梯度洗脱剂b为含0.5mol/l氯化钠的ph7.4、0.01mol/l的tris-hcl缓冲液300ml,线性流速60cm/h,依据280nm波长紫外检测图谱收集各峰,共收集8个层析峰。活性测定和凝胶电泳跟踪确定第6峰为x因子激活剂活性主峰。
2-3)样品透析:步骤2-2)收集的活性峰用截流分子量10000da的膜包(购自赛多利斯,聚醚砜材质)对ph5.0、0.1mol/l的醋酸钠缓冲液进行透析,样品定容至约100ml。置4℃冰箱中备用。
3、sp-sepharosefastflow柱层析:2.6×20cm(填料购自ge公司)
3-1)sp-sepharosefastflow装柱:取保存在20%乙醇中的sp-sepharosefastflow填料约90ml,先用纯净水置换除醇后装填2.6×20cm层析柱,然后用ph5.0、0.1mol/l的醋酸钠缓冲液平衡10个柱体积以上,线性流速100cm/h,280nm波长紫外吸收检测。
3-2)上样、洗脱:将步骤2中透析后的蛇毒溶液泵入已平衡好的层析柱,线性流速60cm/h,上样结束后,用ph5.0、0.1mol/l的醋酸缓冲液进行平衡洗脱,待紫外吸收降至基线后,进行线性梯度洗脱,梯度洗脱剂a为ph5.0、0.1mol/l的醋酸钠缓冲液300ml,梯度洗脱剂b为含0.6mol/l氯化钠的ph5.0、0.1mol/l的醋酸钠缓冲液300ml,线性流速60cm/h,依据280nm波长紫外检测图谱收集各峰,共收集4个层析峰。活性测定和凝胶电泳跟踪确定第3峰为ⅹ因子激活剂活性主峰。
3-3)样品透析:步骤3-2)收集的活性峰用截流分子量10000da的膜包(购自赛多利斯,聚醚砜材质)对含0.3mol/l氯化钠的ph8.0、0.03mol/l的磷酸盐缓冲液进行透析,样品定容至约80ml。置4℃冰箱中备用。
4、cht陶瓷羟基磷灰石i柱层析:1.6×20cm(购自伯乐公司)
4-1)cht陶瓷羟基磷灰石i装柱:取cht陶瓷羟基磷灰石i干粉,用含0.3mol/lnacl的ph8.0、0.03mol/l的磷酸盐缓冲液浸泡4h后取约40ml填料,装填1.6×20cm层析柱,然后用含0.3mol/lnacl的ph8.0、0.03mol/l的磷酸盐缓冲液平衡10个柱体积以上,线性流速100cm/h,280nm波长紫外吸收检测。
4-2)上样、洗脱:将步骤3中透析后的蛇毒溶液泵入已平衡好的层析柱,线性流速80cm/h,上样结束后,用含0.3mol/lnacl的ph8.0、0.03mol/l的磷酸盐缓冲液进行平衡洗脱,待紫外吸收将至基线后,进行线性梯度洗脱,梯度洗脱剂a为含0.3mol/l氯化钠的ph8.0、0.03mol/l的磷酸盐缓冲液150ml,梯度洗脱剂b为含0.8mol/l氯化钠的ph8.0、0.03mol/l的磷酸盐缓冲液150ml,线性流速80cm/h,依据280nm下紫外分光广度检测图谱收集各峰,共收集3个层析峰。活性测定和凝胶电泳跟踪确定第2峰为ⅹ因子激活剂活性主峰。
4-3)样品透析:步骤4-2)收集的活性峰用截流分子量10000da的膜包(购自赛多利斯,聚醚砜材质)对含0.05mol/l氯化钠ph7.4、0.08mol/l的tris-hcl缓冲液进行透析,样品定容至约100ml。置4℃冰箱中备用。
5、deae-sepharosefastflow柱层析:1.6×20cm(填料购自ge公司)
5-1)deae-sepharosefastflow装柱:取保存在20%乙醇中的deae-sepharosefastflow填料约40ml,先用纯净水置换除醇后装填1.6×20cm层析柱,然后用含0.05mol/l氯化钠ph7.4、0.08mol/l的tris-hcl缓冲液平衡10个柱体积以上,线性流速100cm/h,280nm波长紫外吸收检测。
5-2)上样、洗脱:将步骤4中透析后的蛇毒溶液泵入已平衡好的层析柱,线性流速80cm/h小时,上样结束后,用含0.05mol/l氯化钠ph7.4、0.08mol/l的tris-hcl缓冲液进行平衡洗脱,待紫外吸收将至基线后,进行线性梯度洗脱,梯度洗脱剂a为含0.05mol/l氯化钠的、ph7.4、0.08mol/l的tris-hcl缓冲液150ml,梯度洗脱剂b为含0.5mol/l氯化钠的ph7.4、0.08mol/l的tris-hcl缓冲液150ml,线性流速80cm/h,依据280nm波长紫外检测图谱收集各峰,共收集2个层析峰。活性测定和凝胶电泳跟踪确定第2峰为ⅹ因子激活剂活性主峰。
5-3)样品透析:步骤5-2)收集的活性峰用截流分子量10000da的膜包(购自赛多利斯,聚醚砜材质)对含0.1mol/l氯化钠ph7.5、0.05mol/l的tris-hcl缓冲液进行透析浓缩,样品定容至约50ml。置4℃冰箱中备用。
6、superdexg75柱层析:2.6×70cm(填料购自ge公司)
6-1)superdexg75装柱:取保存在20%乙醇中的superdexg75填料约350ml,先用纯净水置换除醇后装填2.6×70cm层析柱,然后用含0.1mol/l氯化钠ph7.5、0.05mol/l的tris-hcl缓冲液平衡5个柱体积以上,线性流速50cm/h,280nm波长紫外吸收检测。
6-2)上样、洗脱:将步骤5中透析浓缩蛇毒溶液,用注射器吸取10ml样品液上样,沿柱侧壁旋转缓慢加入,待样品液完全进入填料后,用少于0.2ml的含0.1mol/l氯化钠的ph7.5、0.05mol/l的tris-hcl缓冲液润洗侧壁和胶面后,加入10ml缓冲液后进行洗脱,线性流速40cm/h。依据280nm波长紫外检测图谱收集各峰,共收集1个层析峰。
7、结果分析:
7-1)纯度检测
采用高效液相色谱(岛津高效液相色谱仪20a,sec-hplc:g2000wxl;rp-hplc:c4色谱柱)和电泳系统进行检测,sec-hplc检测呈单一峰,rp-hplc检测呈单一峰,按照面积归一化法计算,凝血因子ⅹ激活剂含量分别为为100%和95.7%。sds-page变性非还原电泳检测目标蛋白组分显示单一条带,(如图4所示)。
7-2)收率及比活
目标蛋白活性检测经s2765底物法检测,10g溶解后溶液中凝血因子ⅹ激活剂总活性为65万单位,经层析纯化后收集的活性为11.5万单位,收率为17.7%。比活为4282u/mg。
实施例3矛头蝮蛇蛇毒凝血因子ⅹ激活剂的分离纯化
1、蛇毒溶解:
取10g矛头蝮蛇蛇毒,用100mlph8.5、0.1mol/l的tris-hcl缓冲液于4-8℃的层析柜中溶解后,3000g离心15min,取上清液备用。
2、deae-sepharosefastflow柱层析:2.6×20cm(填料购自ge公司)
2-1)deae-sepharosefastflow装柱:取保存在20%乙醇中的deae-sepharosefastflow填料约90ml,先用纯净水置换除醇后装填2.6×20cm层析柱,然后用ph8.5、0.1mol/l的tris-hcl缓冲液平衡10个柱体积以上,线性流速100cm/h,280nm波长紫外吸收检测。
2-2)上样、洗脱:将步骤2-1)中离心后的蛇毒上清液泵入已平衡好的层析柱,线性流速60cm/h,上样结束后,用ph8.5、0.1mol/l的tris-hcl缓冲液进行平衡洗脱,待紫外吸收降至基线后,进行线性梯度洗脱,梯度洗脱剂a为ph8.5、0.1mol/l的tris-hcl缓冲液300ml,梯度洗脱剂b为含0.5mol/l氯化钠的ph8.5、0.1mol/l的tris-hcl缓冲液300ml,线性流速60cm/h,依据280nm波长紫外检测图谱收集各峰,共收集7个层析峰。活性测定和凝胶电泳跟踪确定第6峰为x因子激活剂活性主峰。
2-3)样品透析:步骤2-2)收集的活性峰用截流分子量10000da的膜包对ph6.8、0.02mol/l的磷酸盐缓冲液进行透析,样品定容至约100ml。置4℃冰箱中备用。
3、sp-sepharosefastflow柱层析:2.6×20cm(填料购自ge公司)
3-1)sp-sepharosefastflow装柱:取保存在20%乙醇中的sp-sepharosefastflow填料约90ml,先用纯净水置换除醇后装填2.6×20cm层析柱,然后用ph6.8、0.02mol/l的磷酸盐缓冲液平衡10个柱体积以上,线性流速100cm/h,280nm波长紫外吸收检测。
3-2)上样、洗脱:将步骤2中透析后的蛇毒溶液泵入已平衡好的层析柱,线性流速60cm/h,上样结束后,用ph6.8、0.02mol/l的磷酸盐缓冲液进行平衡洗脱,待紫外吸收降至基线后,进行线性梯度洗脱,梯度洗脱剂a为ph6.8、0.02mol/l的磷酸盐缓冲液300ml,梯度洗脱剂b为含0.5mol/l氯化钠的ph6.8、0.02mol/l的磷酸盐缓冲液300ml,线性流速60cm/h,依据280nm波长紫外检测图谱收集各峰,共收集4个层析峰。活性测定和凝胶电泳跟踪确定第3峰为ⅹ因子激活剂活性主峰。
3-3)样品透析:步骤3-2)收集的活性峰用截流分子量10000da的膜包对含0.3mol/lnacl、0.005mol/lcacl2和mncl2的、ph为7.5、浓度为0.05mol/l的磷酸盐缓冲液进行透析,样品定容至约80ml。置4℃冰箱中备用。
4、conasepharose4b柱层析:1.6×20cm(填料购自ge公司)
4-1)conasepharose4b装柱:取保存在20%乙醇中的conasepharose4b填料约40ml,先用纯净水置换除醇后装填1.6×20cm层析柱,然后用含0.3mol/lnacl、0.005mol/lcacl2和mncl2的、ph为7.5、浓度为0.05mol/l的磷酸盐缓冲液平衡10个柱体积以上,线性流速60cm/h,280nm波长紫外吸收检测。
4-2)上样、洗脱:将步骤3中透析后的蛇毒溶液泵入已平衡好的层析柱,线性流速40cm/h,上样结束后,用ph7.5、0.05mol/l的磷酸盐缓冲液平衡洗脱,待紫外吸收降至基线后,进行线性梯度洗脱,梯度洗脱剂a为ph7.5、0.05mol/l的磷酸盐缓冲液150ml,梯度洗脱剂b为含1mol/l氯化钠的ph7.5、0.05mol/l的磷酸盐缓冲液150ml,线性流速40cm/h,依据280nm波长紫外检测图谱收集各峰,共收集3个层析峰。活性测定和凝胶电泳跟踪确定第2峰为x因子激活剂活性主峰。
4-3)样品透析:步骤4-2)收集的活性峰用截流分子量10000da的膜包对含0.1mol/l氯化钠ph8.5、0.1mol/l的tris-hcl缓冲液进行透析,样品定容至约100ml。置4℃冰箱中备用。
5、deae-sepharosefastflow柱层析:1.6×20cm(填料购自ge公司)
5-3)deae-sepharosefastflow装柱:取保存在20%乙醇中的deae-sepharosefastflow填料约40ml,先用纯净水置换除醇后装填1.6×20cm层析柱,然后用含0.1mol/l氯化钠ph8.5、0.1mol/l的tris-hcl缓冲液平衡10个柱体积以上,线性流速100cm/h小时,280nm波长紫外吸收检测。
5-2)上样、洗脱:将步骤4中透析后的蛇毒溶液泵入已平衡好的层析柱,线性流速60cm/h,上样结束后,用含0.1mol/l氯化钠ph8.5、0.1mol/l的tris-hcl缓冲液进行平衡洗脱,待紫外吸收降至基线后,进行线性梯度洗脱,梯度洗脱剂a为含0.1mol/l氯化钠ph8.5、0.0mol/l的tris-hcl缓冲液150ml,梯度洗脱剂b为含0.5mol/l氯化钠的ph8.5、0.1mol/l的tris-hcl缓冲液150ml,线性流速60cm/h,依据280nm波长紫外检测图谱收集各峰,共收集2个层析峰。活性测定和凝胶电泳跟踪确定第1峰为x因子激活剂活性主峰。
5-3)样品透析:步骤5-2)收集的活性峰用截流分子量10000da的膜包对ph8.0、0.05mol/l的磷酸盐缓冲液进行透析浓缩,样品定容至约50ml。置4℃冰箱中备用。
6、superdexg75柱层析:2.6×70cm(填料购自ge公司)
6-1)superdexg75装柱:取保存在20%乙醇中的superdexg75填料约350ml,先用纯净水置换除醇后装填2.6×70cm层析柱,然后用ph8.0、0.05mol/l的磷酸盐缓冲液平衡5个柱体积以上,线性流速50cm/h,280nm波长紫外吸收检测。
6-2)上样、洗脱:将步骤5中透析浓缩蛇毒溶液,用注射器吸取10ml样品液上样,沿柱侧壁旋转缓慢加入,待样品液完全进入填料后,用少于0.2ml的ph8.0、0.05mol/l的磷酸盐缓冲液润洗侧壁和胶面后,加入10ml缓冲液后进行洗脱,线性流速40cm/h。依据280nm下紫外分光广度检测图谱收集各峰,共收集1个层析峰。
7、结果分析:
7-1)纯度检测
采用高效液相色谱(岛津高效液相色谱仪20a,sec-hplc:g2000wxl;rp-hplc:c4色谱柱)和电泳系统进行检测,sec-hplc检测呈单一峰,rp-hplc检测呈单一峰,按照面积归一化法计算,凝血因子ⅹ激活剂含量分别为为100%和94.7%。sds-page变性非还原电泳检测目标蛋白组分显示单一条带。
7-2)收率及比活
目标蛋白活性检测经s2765底物法检测,10g溶解后溶液中凝血因子ⅹ激活剂总活性为63万单位,经层析纯化后收集的活性为12.2万单位,收率为19.4%。比活为4260u/mg。
实施例4矛头蝮蛇蛇毒凝血因子ⅹ激活剂功能研究
本研究考察矛头蝮蛇凝血因子x激活剂促凝作用。
实验一
1.实验目的
考察矛头蝮蛇凝血因子x激活剂对人正常血浆和乏viii因子、乏ix因子血浆活化部分凝血活酶时间(aptt)的作用。
2.试剂及仪器
正常凝血质控血浆德国美创公司批号047b-d087a
乏viii因子血浆西门子公司批号503627
乏ix因子血浆西门子公司批号500874d
aptt试剂盒德国美创公司批号11197753
fxa原料实施例1纯化的fxa用生理盐水稀释到一定浓度
coatronm4半自动凝血分析仪,德国teco
3.实验方法
3.1fxa原料对正常凝血质控血浆appt的作用
参照aptt测试试剂盒方法,稍作修改。正常凝血质控血浆,放至室温,每支加入1.0ml去离子水溶解,室温放置15min,期间间或轻摇,备用。取不同浓度的fxa溶液0.01ml,加入0.09ml备用的正常凝血质控血浆,混匀,37℃预热2min,加入已37℃预热的aptt试剂0.1ml,继续保温3min,加入37℃预热的氯化钙0.1ml,记录血浆的aptt时间。以去离子水代替fxa溶液作为空白对照。
3.2fxa原料对乏因子血浆aptt的作用
参照aptt测试试剂盒方法,稍作修改。乏因子血浆,放至室温,每支加入1.0ml去离子水溶解,室温放置15min,期间间或轻摇,备用。取不同浓度的fxa溶液0.01ml,加入0.09ml备用乏因子血浆,混匀,37℃保温2min,加入已37℃预热的aptt试剂0.1ml,继续保温3min,加入37℃预热的氯化钙0.1ml,记录血浆的aptt时间。空白对照组加入等体积的去离子水。
4.实验结果
4.1fxa原料对人正常血浆aptt的作用
不同浓度fxa原料对人正常血浆aptt时间的作用如表1所示。
与空白对照组比较,0.1,0.075,0.05,0.025,0.01u/ml的fxa均能显著性缩短人正常血浆的aptt,并表现出剂量依赖性(p<0.05)。
表1fxa原料对人正常血浆aptt的作用
以正常血浆为空白对照组(con),给予不同浓度的fxa,比较aptt的时间。***p<0.001,与空白对照组比较;*p<0.05,与空白对照组比较(n=4)。
4.2fxa原料对人乏viii因子血浆aptt的作用
不同浓度fxa对人乏viii因子血浆aptt的作用如表2。与人正常血浆比较,乏viii因子血浆组aptt时间显著延长(p<0.001)。在给予不同浓度的fxa后,乏viii因子血浆的aptt显著性缩短,并表现出剂量依赖性(p<0.001)。其中,0.1u/mlfxa组的aptt,与正常血浆凝血时间相近。
表2fxa原料对人乏viii因子血浆aptt的作用
以正常血浆为空白对照组(con),乏viii血浆组(viiifree),不同浓度的fxa组,比较aptt的时间。***p<0.001,与空白对照组比较;###p<0.001,与乏viii因子血浆组比较(n=4)。
4.3fxa原料对人乏ix因子血浆aptt的作用
不同浓度fxa对人乏ix因子血浆aptt的作用如表3。与人正常血浆比较,乏ix因子血浆组aptt时间显著延长(p<0.001)。在给予不同浓度的fxa后,乏ix因子血浆的aptt显著性缩短,并表现出剂量依赖性(p<0.01)。其中,0.1u/mlfxa组的aptt,与正常血浆凝血时间相近。
表3fxa原料对人乏ix因子血浆aptt的作用
以正常血浆为空白对照组(con),乏ix血浆组(ixfree),不同浓度的fxa组,比较aptt的时间。***p<0.001,与空白对照组比较;###p<0.001,##p<0.01,与乏ix因子血浆组比较(n=4)。
5.结论
体外实验中,凝血因子x激活剂可以明显缩短正常血浆和乏viii因子、乏ix因子血浆活化部分凝血活酶时间(aptt)时间,并表现出剂量依赖性。
对比实施例5从圆斑蝰蛇蛇毒中提取凝血因子x激活剂
1、蛇毒溶解:
10g圆斑蝰蛇蛇毒(购自辽宁远大诺康生物制药有限公司),用100mlph8.0、0.05mol/l的tris-hcl缓冲液于4-8℃的层析柜中溶解后,3000g离心15min,取上清液备用。
2、deae-sepharosefastflow柱层析:2.6×20cm(填料购自ge公司)
2-1)deae-sepharosefastflow装柱:取保存在20%乙醇中的deae-sepharosefastflow填料约90ml,先用纯净水置换除醇后装填2.6×20cm层析柱,然后用ph8.0、0.05mol/l的tris-hcl缓冲液平衡10个柱体积以上,线性流速100cm/h,280nm波长紫外吸收检测。
2-2)上样、洗脱:将步骤2-1)中离心后的蛇毒上清液泵入已平衡好的层析柱,线性流速60cm/h,上样结束后,用ph8.0、0.05mol/l的tris-hcl缓冲液进行平衡洗脱,待紫外吸收降至基线后,进行线性梯度洗脱,梯度洗脱剂a为ph8.0、0.05mol/l的tris-hcl缓冲液300ml,梯度洗脱剂b为含0.5mol/l氯化钠的ph8.0、0.05mol/l的tris-hcl缓冲液300ml,线性流速60cm/h,依据280nm波长紫外检测图谱收集各峰,共收集6个层析峰。活性测定和凝胶电泳跟踪确定第3峰为ⅹ因子激活剂活性主峰。
2-3)样品透析:步骤2-2)收集的活性峰用截流分子量10000da的膜包对ph6.0、0.05mol/l的磷酸盐缓冲液进行透析,样品定容至约100ml。置4℃冰箱中备用。
3、sp-sepharosefastflow柱层析:2.6×20cm(填料购自ge公司)
3-1)sp-sepharosefastflow装柱:取保存在20%乙醇中的sp-sepharosefastflow填料约90ml,先用纯净水置换除醇后装填2.6×20cm层析柱,然后用ph6.0、0.05mol/l的磷酸盐缓冲液平衡10个柱体积以上,线性流速100cm/h,280nm波长紫外吸收检测。
3-2)上样、洗脱:将步骤2中透析后的蛇毒溶液泵入已平衡好的层析柱,线性流速60cm/h,上样结束后,用ph6.0、0.05mol/l的磷酸盐缓冲液进行平衡洗脱,待紫外吸收降至基线后,进行线性梯度洗脱,梯度洗脱剂a为ph6.0、0.05mol/l的磷酸盐缓冲液300ml,梯度洗脱剂b为含0.5mol/l氯化钠的ph6.0、0.05mol/l的磷酸盐缓冲液300ml,线性流速60cm/h,依据280nm波长紫外检测图谱收集各峰,共收集5个层析峰。活性测定和凝胶电泳跟踪确定第3峰为x因子激活剂活性主峰。
3-3)样品透析:步骤3-2)收集的活性峰用截流分子量10000da的膜包对含0.8mol/l氯化钠、0.002mol/l氯化钙和0.002mol/l氯化锰的ph8.0、0.03mol/l的tris-hcl缓冲液进行透析,样品定容至约80ml。置4℃冰箱中备用。
4、lentillectinsepharose4b柱层析:1.6×20cm(填料购自ge公司)
4-1)lentillectinsepharose4b装柱:取保存在20%乙醇中的lentillectinsepharose4b填料约40ml,先用纯净水置换除醇后装填1.6×20cm层析柱,然后用含0.8mol/l氯化钠、0.002mol/l氯化钙和0.002mol/l氯化锰的ph8.0、0.03mol/l的tris-hcl缓冲液平衡10个柱体积以上,线性流速60cm/h,280nm波长紫外吸收检测。
4-2)上样、洗脱:将步骤3中透析后的蛇毒溶液泵入已平衡好的层析柱,线性流速40cm/h,上样结束后,用含0.8mol/l氯化钠、0.002mol/l氯化钙和0.002mol/l氯化锰的ph8.0、0.03mol/l的tris-hcl缓冲液平衡洗脱,待紫外吸收降至基线后,进行线性梯度洗脱,梯度洗脱剂a为含0.8mol/l氯化钠的ph8.0、0.03mol/l的tris-hcl缓冲液150ml,梯度洗脱剂b为含0.8mol/l氯化钠和0.4mol/l甲基-α-d-吡喃葡萄糖苷的ph8.0、0.03mol/l的tris-hcl缓冲液150ml,线性流速40cm/h,依据280nm波长紫外检测图谱收集各峰,共收集3个层析峰。活性测定和凝胶电泳跟踪确定穿透峰为x因子激活剂活性主峰。
4-3)样品透析:步骤4-2)收集的活性峰用截流分子量10000da的膜包对含0.1mol/l氯化钠ph8.0、0.03mol/l的tris-hcl缓冲液进行透析,样品定容至约100ml。置4℃冰箱中备用。
5、deae-sepharosefastflow柱层析:1.6×20cm(填料购自ge公司)
5-1)deae-sepharosefastflow装柱:取保存在20%乙醇中的deae-sepharosefastflow填料约40ml,先用纯净水置换除醇后装填1.6×20cm层析柱,然后用含0.1mol/l氯化钠ph8.0、0.03mol/l的tris-hcl缓冲液平衡10个柱体积以上,线性流速100cm/h,280nm波长紫外吸收检测。
5-2)上样、洗脱:将步骤4中透析后的蛇毒溶液泵入已平衡好的层析柱,线性流速60cm/h小时,上样结束后,用含0.1mol/l氯化钠ph8.0、0.03mol/l的tris-hcl缓冲液进行平衡洗脱,待紫外吸收将至基线后,进行线性梯度洗脱,梯度洗脱剂a为含0.1mol/l氯化钠ph8.0、0.03mol/l的tris-hcl缓冲液150ml,梯度洗脱剂b为含0.5mol/l氯化钠的ph8.0、0.03mol/l的tris-hcl缓冲液150ml,线性流速60cm/h,依据280nm波长紫外检测图谱收集各峰,共收集3个层析峰。活性测定和凝胶电泳跟踪确定第2峰为ⅹ因子激活剂活性主峰。
5-3)样品透析:步骤5-2)收集的活性峰用截流分子量10000da的膜包对含0.05mol/l氯化钠ph8.0、0.02mol/l的tris-hcl缓冲液进行透析浓缩,样品定容至约50ml。置4℃冰箱中备用。
6、superdexg75柱层析:2.6×70cm(填料购自ge公司)
6-1)superdexg75装柱:取保存在20%乙醇中的superdexg75填料约350ml,先用纯净水置换除醇后装填2.6×70cm层析柱,然后用含0.05mol/l氯化钠的ph8.0、0.02mol/l的tris-hcl缓冲液平衡5个柱体积以上,线性流速60cm/h小时,280nm波长紫外吸收检测。
6-2)上样、洗脱:将步骤5中透析浓缩蛇毒溶液,用注射器吸取10ml样品液上样,沿柱侧壁旋转缓慢加入,待样品液完全进入填料后,用少于0.2ml的含0.05mol/l氯化钠的ph8.0、0.02mol/l的tris-hcl缓冲液润洗侧壁和胶面后,加入10ml缓冲液后进行洗脱,线性流速40cm/h。依据280nm下紫外分光广度检测图谱收集各峰,共收集1个层析峰。
7、结果分析:
7-1)纯度检测
采用高效液相色谱(岛津高效液相色谱仪20a,sec-hplc:g2000wxl;rp-hplc:c4色谱柱)和电泳系统进行检测,sec-hplc检测呈2个峰,rp-hplc检测呈2峰,按照面积归一化法计算,凝血因子ⅹ激活剂含量分别为为82%和57%。sds-page变性非还原电泳检测目标蛋白组分显示多条谱带。
7-2)收率及比活
目标蛋白活性检测经s2765底物法检测,10g溶解后溶液中凝血因子ⅹ激活剂总活性为70万单位,经层析纯化后收集的活性为8万单位,收率约为11.4%。比活为987u/mg。
8.结论
用本发明的方法从圆斑蝰蛇蛇毒中提取凝血因子ⅹ激活剂,收率仅为11.4%,纯度和比活均较低,说明本专利方法特别适合于矛头蝮蛇蛇毒凝血因子ⅹ激活剂的提取,同时,适用本发明的方法提取的矛头蝮蛇蛇毒凝血因子ⅹ激活剂含量较高,更适合规模化的生产。
对比实施例6现有方法从矛头蝮蛇蛇毒提取凝血因子ⅹ激活剂
提取步骤:(参照hofmann,h.,dumarey,c.,bon,c.(1983).bloodcoagulationinducedbybothropsatroxvenom.identificationandpropertiesofafactorⅹactivator.biochimie65(3):201–210)。)
1、蛇毒溶液制备:
1.5g矛头蝮蛇蛇毒,用30ml含ph7.5、0.05mol/l的磷酸盐缓冲液于4℃的层析柜中溶解后,8000rpm离心30min,取上清液用相同的缓冲液进行透析,定容40ml备用。
2、deae-cellulose柱层析:2.5×60cm(de52填料购自ge公司)
2-1)deae-cellulose柱前平衡:220ml填料装柱,用ph7.5、0.05mol/l的磷酸盐缓冲液平衡10个柱体积以上。
2-2)上样、洗脱、收集:约40ml蛇毒上清液泵入已平衡好的层析柱,0.5ml/min,洗脱过程分三步,500ml平衡液洗脱→1000ml含0-0.3mol/l氯化钠的ph7.5、0.05mol/l的磷酸盐缓冲液梯度洗脱→含1mol/l氯化钠的ph7.5、0.05mol/l的磷酸盐缓冲液直线洗脱。依据280nm波长紫外检测图谱收集各峰,共收集7个层析峰。s2765底物法活性测定和凝胶电泳跟踪确定第4峰为ⅹ因子激活剂活性主峰。
2-3)样品冻干透析:步骤2-2)收集的活性峰进行冻干,用10mlph7.5、5mmol/ltris-hcl缓冲液回溶,8000g离心15min除沉淀,再对回溶液进行透析,膜包截流分子量为10000da,定容30ml后进行冻干备用。
3、sephadexg100(superfine)柱层析:2×100cm(填料购自ge)
3-1)sephadexg100柱平衡:
300ml填料装柱,ph7.5、5mmol/l的tris-hcl缓冲液平衡10cv以上。流速0.5ml/min。
3-2)上样、洗脱:将步骤2-3)中冻干品用5mlph7.5、5mmol/l的tris-hcl缓冲液回溶,用注射器吸取样品液5ml上样,沿柱侧壁旋转缓慢加入,待样品液完全进入填料后,用少于0.2ml缓冲液润洗侧壁和胶面后进行洗脱,流速0.5ml/min,依据280nm下紫外分光广度检测图谱收集各峰,共收集3个层析峰。s2765底物法活性测定和凝胶电泳跟踪确定第2峰为ⅹ因子激活剂活性主峰。
7、结果分析:
7-1)纯度检测
采用高效液相色谱(岛津高效液相色谱仪20a,sec-hplc:g2000wxl;rp-hplc:c4色谱柱)和电泳系统进行检测,sec-hplc检测呈单一峰,rp-hplc检测呈单一峰,按照面积归一化法计算,凝血因子ⅹ激活剂含量分别为为76%和53%。sds-page变性非还原电泳检测目标蛋白组分显示4条带。
7-2)收率及比活
目标蛋白活性检测经s2765底物法检测,1.5g溶解后溶液中凝血因子ⅹ激活剂总活性为9万单位,经层析纯化后收集的活性为2.25万单位,收率为25%。比活为876u/mg。
7、结论:
从矛头蝮蛇蛇毒中分离凝血因子ⅹ激活剂的报道较少,国内文献及专利报道多是从圆斑蝰蛇蛇毒中提取凝血因子ⅹ激活剂,实验人参照文献方法从矛头蝮蛇蛇毒中提取凝血因子ⅹ激活剂,收率较高为25%,但纯度和比活均较低,说明文献的方法不适用于规模化制备高纯度的凝血因子ⅹ激活剂。
序列表
<110>辽宁远大诺康生物制药有限公司
<120>一种凝血因子x激活剂及其制备方法
<130>dic16110057
<160>3
<170>patentinversion3.5
<210>1
<211>15
<212>prt
<213>人工序列
<220>
<223>凝血因子ⅹ激活剂的h链n-末端序列
<400>1
leuthrprogluglnglnalatyrleuaspalalyslystyrval
151015
<210>2
<211>15
<212>prt
<213>人工序列
<220>
<223>凝血因子ⅹ激活剂的l1链n-末端
<400>2
aspcysproserasptrpsersertyrgluglyhiscystyrarg
151015
<210>3
<211>15
<212>prt
<213>人工序列
<220>
<223>凝血因子ⅹ激活剂的l2链n-末端
<400>3
aspcysasnsertyrtrpserthrtyrgluglyargsertyrarg
151015
- 还没有人留言评论。精彩留言会获得点赞!