一种尾接有机导向分子的芳基钌配合物及其合成方法和应用与流程
本发明涉及一种尾接有机导向分子的芳基钌配合物,具体涉及一种以18β-甘草次酸功能团为有机导向分子的芳基钌金属配合物,还涉及上述芳基钌金属配合物的合成方法和应用。
技术背景
金属抗癌药物由于其独特的药理作用而被广泛应用于药物化学的研究,其中临床使用较多的抗肿瘤化学药物为顺铂、卡铂以及奥沙利铂,但铂类药物存在效率低、毒副作用大、选择性差以及耐药等缺点,因此,研究开发新型高效、低毒的抗肿瘤药物一直备受世界瞩目。自上世纪七十年代开始,芳基钌化合物作为抗癌活性物质的研究已取得重大进展,部分芳基钌化合物的抗癌活性与目前临床治疗效果最好的顺铂相当,而在未来芳基钌抗癌化合物的结构设计上,许多科学家都认为构建具有靶向性能的芳基钌化合物可以赋予其高效选择性的优点。在这方面,英国华威大学的petersadler课题组将含有受体靶向性的多肽连接到芳基钌配合物上,修饰后的配合物具有双重选择性的抗癌机制,该结果发表于j.am.chem.soc期刊。由此可见,构建具有靶向性的芳基钌抗癌配合物将成为未来设计金属抗癌药物重要的研究策略。
技术实现要素:
本发明所要解决的技术问题是提供一种尾接有机导向分子的芳基钌配合物,该芳基钌配合物尾接有18β-甘草次酸功能团作为导向分子,从而使其(芳基钌配合物)具有良好的靶向性。
本发明还要解决的技术问题是提供上述尾接有机导向分子的芳基钌配合物的合成方法,该方法工艺流程简单、易于操作,且产率高。
本发明最后要解决的技术问题是提供上述尾接有机导向分子的芳基钌配合物在用于制备抗癌药物、抗癌药物组分、抗菌药物以及抗菌药物组分方面的应用。
为解决上述技术问题,本发明所采用的技术方案为:
一种尾接有机导向分子的芳基钌配合物,具有如下结构式:
上述尾接有机导向分子的芳基钌配合物的合成方法,包括如下步骤:
步骤1,用甘草次酸合成具有18β-甘草次酸功能团的有机化合物;
步骤2,在惰性氛围下,将所需量的对伞花烃芳基钌二聚体(对伞花烃二氯化钌二聚体)与步骤1制得的有机化合物溶解于溶剂中,加热反应后加入乙醚,最后离心得到所需产物。
其中,步骤1中,具有18β-甘草次酸功能团有机化合物的合成方法为:
在惰性氛围下,将所需量的18β-甘草次酸与桥联配体2-(1-咪唑基)乙胺或4-(4'-甲基-2,2'-联吡啶)-甲胺溶解于有机溶剂中,有机溶剂中还含有一定量的1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐和1-羟基苯并三唑;混合物料于一定温度下反应一段时间后得到粗产物,将粗产物经过柱层析法分离纯化后即可得到具有18β-甘草次酸功能团有机化合物。
其中,步骤1中,具有18β-甘草次酸功能团有机化合物的结构式为:
其中,步骤2中,所述对伞花烃芳基钌二聚体与有机化合物的反应摩尔比为5:1~1:20。
其中,步骤2中,所述溶剂为二氯甲烷、氯仿、甲醇、乙醇或乙二醇;反应温度为2~90℃。
其中,所述18β-甘草次酸与桥联配体2-(1-咪唑基)乙胺或4-(4'-甲基-2,2'-联吡啶)-甲胺的反应摩尔比为5:1~1:10。合成有机化合物的方法中,edci(1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐)用于羧基的活化试剂,hobt(1-羟基苯并三唑)用于缩合反应的催化剂。
其中,所述有机溶剂为n,n-二甲基甲酰胺或其他甲酰胺、乙酰胺溶剂;反应温度为-20~120℃。
上述尾接有机导向分子的芳基钌配合物在用于制备抗癌药物、抗癌药物组分、抗菌药物以及抗菌药物组分方面的应用。
上述尾接有机导向分子的芳基钌配合物的合成方法中具有18β-甘草次酸功能团有机化合物在用于制备抗癌药物以及抗癌药物组分方面的应用。
本发明尾接有机导向分子的芳基钌配合物的合成方法中,桥联配体2-(1-咪唑基)乙胺的制备方法如下:步骤(1)、将所需量的咪唑、丙烯酸乙酯和fecl3·6h2o溶于水中,反应0.1~15h后用二氯甲烷萃取,减压蒸去溶剂,粗产物通过柱层析法分离纯化,得淡黄色液体;步骤(2)、将水合肼和乙醇混合,加热条件下加入乙醇与步骤(1)产物的混合物,加热回流反应0.2~9h后用二氯甲烷萃取,减压蒸去溶剂,粗产物通过柱层析法分离纯化,得黄色油状物;步骤(3)、将步骤(2)中的产物、水和浓硫酸加入到三颈瓶中,低温条件下缓慢滴加到含有nano2的溶液中,反应0.1~3h后再于加热条件下反应0.3~6h,冷却至室温,加氢氧化钠水溶液调ph至7~11,减压蒸去溶剂,向剩余物中加入甲醇,抽滤,滤液蒸去溶剂,粗产物通过柱层析法分离纯化,得淡黄色油状物即为2-(1-咪唑基)乙胺。
本发明尾接有机导向分子的芳基钌配合物的合成方法中,桥联配体4-(4'-甲基-2,2'-联吡啶)-甲胺制备方法如下:步骤(1)、将seo2(二氧化硒)、1,4-dioxide(1,4-二氧六环)和4,4'-dimethyl-2,2'-bipyridyl(4,4'-二甲基-2,2'-联吡啶)混合于三颈瓶中,回流加热反应0.4~15h,反应结束后,冷却过滤,旋转蒸发溶剂,残留物中加入nahco3溶液,用二氯甲烷萃取后旋蒸除去溶剂,残留物中加入一定浓度的na2s2o5,过滤,滤液调ph至8~12,再用二氯甲烷萃取,减压蒸去溶剂,产物为固体粉末;步骤(2)、将盐酸羟胺、k2co3以及步骤(1)的产物溶于甲醇与水的混合溶液中,加热条件下反应一段时间,反应结束后冷却至室温,加入适量的水,过滤,得固体粉末;步骤(3)、将步骤(2)的产物、醋酸铵以及氨水溶于乙醇与水的混合溶液中,回流加热一段时间,加入锌粉,反应0.2~4h,冷却至室温,过滤,旋蒸除去溶剂,加入强碱溶液,使用二氯甲烷萃取,减压蒸去溶剂,得到固体粉末即为4-(4'-甲基-2,2'-联吡啶)-甲胺。
本发明具有18β-甘草次酸功能团有机化合物在经桥联配体2-(1-咪唑基)乙胺或4-(4'-甲基-2,2'-联吡啶)-甲胺修饰后可得尾接18β-甘草次酸官能团作为导向分子的芳基钌配合物,该芳基钌配合物具有良好的靶向性和抗癌抗菌活性,通过研究该芳基钌配合物的抗癌抗菌活性发现双齿配位配合物的活性要远远高于单齿配位配合物的活性。
本发明尾接有机导向分子的芳基钌配合物合成方法的反应链为:
与现有技术相比,本发明技术方案具有的有益效果为:
本发明尾接有机导向分子的芳基钌配合物具有良好的靶向性和抗癌抗菌活性,合成该芳基钌配合物的方法工艺流程简单、易于操作,且产率高;本发明尾接有机导向分子的芳基钌配合物能够应用于制备抗癌药物、抗癌药物组分、抗菌药物以及抗菌药物组分。
附图说明
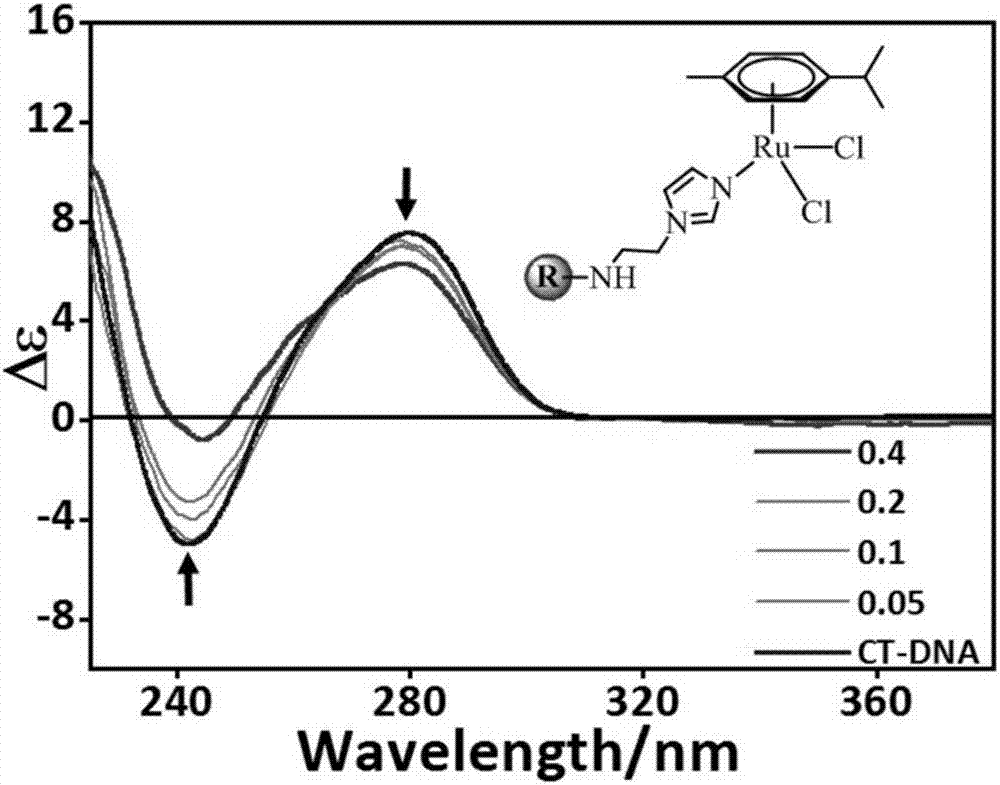
图1是实施例3尾接有机导向分子的芳基钌配合物与dna相互作用的圆二色谱图;
图2是实施例4尾接有机导向分子的芳基钌配合物与dna相互作用的圆二色谱图;
图3是实施例3尾接有机导向分子的芳基钌配合物的电泳图;
图4是实施例4尾接有机导向分子的芳基钌配合物的电泳图;
图5是实施例4尾接有机导向分子的芳基钌配合物促进细胞内活性氧生成的共聚焦荧光成像图;
图6是实施例4尾接有机导向分子的芳基钌配合物的细胞流式图。
具体实施方式
以下结合附图和具体实施例对本发明的技术方案做进一步说明。
实施例1
在氩气氛围下,将18β-甘草次酸(0.22g,0.5mmol),edci(1.05g,5.5mmol),hobt(0.61g,4.5mmol)以及桥联配体2-(1-咪唑基)乙胺(0.56g,5.0mmol)溶解于dmf(n,n-二甲基甲酰胺)中,于20℃下搅拌5h,粗产物通过柱层析法分离纯化,得到经桥联配体修饰后的有机化合物l1,产率为58%。
有机化合物l1的结构式为:
元素分析:理论值(%):c35h53n3o3·(c3h8o):c73.15,h9.85,n6.74;实验值:c73.24,h9.62,n6.66。1hnmr(400hz,cdcl3):δ7.44(s,1h,him),7.10(s,1h,him),6.94(s,1h,him),6.01(s,br,1h,conh),5.61(s,1h,h18β-ga),4.14(m,2h,him),3.61(m,2h,him),3.23(dd,1h,h18β-ga,j1=5.6,j2=10.8hz),2.79(d,1h,h18β-ga,j=13.6hz),2.33-0.69(42h,3ch,9ch2and7ch3of18β-ga)。esi-ms(+):理论值:m/z564.41,实验值:m/z564.83。
实施例2
在氩气氛围下,将18β-甘草次酸(0.22g,0.5mmol),edc(1.05g,5.5mmol),hobt(0.61g,4.5mmol)以及桥联配体4-(4'-甲基-2,2'-联吡啶)-甲胺(1.0g,5.0mmol)溶解于dmf中,于20℃下搅拌5h,粗产物通过柱层析法分离纯化,得到经桥联配体修饰后的有机化合物l2,产率为65%。
有机化合物l2的结构式为:
元素分析:理论值(%)c42h57n3o3·0.5(ch3oh):c76.42,h8.90,n6.29,实验值:c76.04,h9.02,n5.82。1hnmr(400hz,cdcl3):mr(400hz,cdclbpy,j=5.2hz),8.51(d,1h,hbpy,j=4.8hz),8.34(s,1h,hbpy),8.24(s,1h,hbpy),7.23(d,1h,hbpy,j=4.0hz),7.15(d,1h,hbpy,j=4.4hz),6.24(s,br,1h,conh),5.66(s,1h,h18β-ga),4.58(m,2h,hbpy),3.23(dd,1h,h18β-ga,j1=5.6,j2=10.8hz),2.79(d,1h,h18β-ga,j=13.2hz),2.45(s,3h,hbpy),2.33-0.69(42h,3ch,9ch2and7ch3of18ch0。esi-ms(+):理论值:m/z652.44,实验值:m/z652.83。
实施例3
在氩气氛围下,将有机化合物l1(0.07g,0.1mmol)与对伞花烃芳基钌二聚体(对伞花烃二氯化钌二聚体)(0.31g,0.5mmol)溶解于甲醇中,反应结束后(反应温度为65℃),加入乙醚,离心得到具有有机导向分子的单齿配位芳基钌配合物1,产率为87%。
芳基钌配合物1的结构式为:
元素分析:理论值(%)c45h67cl2n3o3ru·2(h2o):c60.86,h7.76,n4.83,实验值:cc60.51,h7.83,n4.73。1hnmr(400hz,cdcl3):δ8.22(s,1h,him),7.24(s,1h,him),6.91(s,1h,him),6.89(s,br,1h,conh),5.73(s,1h,h18β-ga),5.41(m,2h,hbzofp-cymene),5.27(d,2h,hbzofp-cymene,j=6.0hz),3.80(m,2h,him),2.93(m,2h,him),3.22(s,1h,h18β-ga),2.81(d,h18β-ga,j=13.6hz),2.46–0.71(10h,1chand3ch3ofp-cymene),2.34-0.69(m,42h,3ch,9ch2and7ch3of18β-ga).esi-ms(+):理论值:[1-cl-]+m/z:834.36,实验值:m/z834.92。
实施例4
在氩气氛围下,将有机化合物l2(0.06g,0.1mmol)与对伞花烃芳基钌二聚体(对伞花烃二氯化钌二聚体)(0.31g,0.5mmol)溶解于甲醇中,反应结束后(反应温度为65℃)加入乙醚,离心得到具有有机导向分子的双齿配位芳基钌配合物2,产率为85%。
芳基钌配合物2的结构式为:
元素分析:理论值(%)c52h71cl2n3o3ru·3(h2o):c61.71,h7.67,n4.15,实验值:c61.29,h7.58,n4.36。1hnmr(400hz,d6-dmso):δ9.47(d,1h,hbpy,j=5.6hz),9.37(d,1h,hbpy,j=6.0hz),8.51(s,1h,hbpy),8.40(s,1h,hbpy),7.56(d,1h,hbpy,j=5.6hz),7.51(d,1h,hbpy,j=6.4hz),8.49(s,br,1h,conh),6.20(dd,2h,hbzofp-cymene,j1=3.6hz,j2=6.4hz),5.95(dd,2h,hbzofp-cymene,j1=2.0hz,j2=5.2hz),5.49(d,1h,h18β-ga,j=13.2hz),4.52(m,2h,hbpy),4.32(d,1h,h18β-ga),3.02(m,1h,h18β-ga),2.56(s,3h,hbpy),2.54–0.71(10h,1chand3ch3ofp-cymene),2.33-0.69(42h,3ch,9ch2and7ch3of18β-ga)。esi-ms(+):理论值:[2-cl-]+m/z:923.42,实验值:m/z923.08。
本发明具有18β-甘草次酸功能团的有机化合物以及含有机导向分子的芳基钌配合物抗癌活性方面的研究:hela(宫颈癌)、a2780(卵巢癌细胞)以及mcf-7(乳腺癌)。
方法:mtt比色法,测定具有18β-甘草次酸功能团的有机化合物以及尾接有机导向分子的芳基钌配合物对人源癌细胞系(hela(宫颈癌),mcf7(乳腺癌),a2780(卵巢癌))在体外的抗癌活性。将hela,mcf7和a2780细胞置于具有10%胎牛血清和1%青霉素-链霉素溶液的dmem培养基中,37℃,5%co2细胞培养箱中培养。将细胞以5000细胞/孔的初始密度接种到96孔细胞培养板中,在培养24小时后移除培养基,作4组对比试验,4组试验组分别加入不同浓度的有机化合物l1(实施例1)、有机化合物l2(实施例2)、配合物1(实施例3)和配合物2(实施例4),继续孵育44h。之后,在每个试验组孔中加入20μl的mtt溶液,继续孵育4h,最后除去培养基,加入150mldmso,震摇10分钟,并在eliasa(tecaninfinitem1000pro)酶标仪上读取590nm处的吸光度。
实施例1~4制得的有机化合物l1、有机化合物l2、金属配合物1和金属配合物2的抗癌活性如下表所示。
表1为18β-甘草次酸(18β-ga)、有机化合物l1、有机化合物l2、金属配合物1、金属配合物2以及顺铂(cddp)的ic50(μm)值:
结果表明,经过n,n-螯合配体修饰后的甘草次酸相较于未修饰的甘草次酸,其活性获得了很大程度的提高,同时,实施例2制得的双齿有机化合物l2与实施例4制得的双齿配位芳基钌配合物2的活性要远远高于实施例1制得的单齿有机化合物l1以及实施例3制得的单齿配位芳基钌配合物1,这可能是因为双齿配位配合物的稳定性较好,其化学溶解性能较好的与生物环境相匹配,而单齿配位配合物的稳定性较差,且配体中咪唑较好的水溶性也影响了其细胞摄取。
本发明具有有机导向分子芳基钌配合物在抗菌方面的应用:
方法:微量肉汤稀释法,体外测定具有有机导向分子芳基钌配合物对金黄色葡萄球菌以及大肠杆菌的最低杀菌浓度(mbc)以及最低抑菌浓度(mic)。
将对数生长期的菌种接种于普通肉汤培养基中并将其浓度稀释至5×105cfu/ml,将稀释好的等梯度抗菌药物(0.125-256μg/ml)加入到96孔平板中,每孔100μl,然后将提前稀释好的菌液加入到96孔平板中,每孔的最终体积为200μl,如此孔中抗菌药物的最终浓度为0.125-256μg/ml,将96孔平板置于垫有湿脱脂棉的托盘内,不震摇的情况下37℃培养18h。最低抑菌浓度在瑞士帝肯tecan多功能酶标仪平台spark10m上读取600nm处的吸光度。取mic终点以上未长菌的培养液5μl移种于另外的琼脂平板上,将琼脂平板置于37℃的恒温箱中培养18h,则培养后平板上无菌生长的药物最低浓度即是最低杀菌浓度(mbc)。
实施例3~4制得的芳基钌金属配合物1,2的抗菌活性如表2所示。
表2为芳基钌金属配合物1,2的最低抑菌浓度(mic)和最低杀菌浓度(mbc)值。
结果表明,以咪唑基为桥联配体的单齿配位芳基钌配合物1基本无抗菌活性,而以联吡啶基为桥联配体的双齿配位芳基钌配合物2表现出比较显著的抗菌活性,其对革兰氏阳性菌金黄色葡萄球菌以及革兰氏阴性菌大肠杆菌mic/mbc的范围在2.0-8.0μg/ml,配合物2可能是由于引入了脂溶性较好的联吡啶基从而可有效地提高其抗菌活性。
本发明尾接有机导向分子的芳基钌配合物与dna作用的应用:
方法一:圆二色谱法。通过cd光谱在pbs缓冲溶液(5mm,ph=7.2)中研究配合物1,2与ct-dna的相互作用。配制一系列不同摩尔比的(ri=[配合物]/[ct-dna],ri值分别为0,0.05,0.1,0.2的样品,其中,ct-dna碱基对浓度设定为100μm,于37℃恒温箱中孵化24小时。在1cm宽度的比色皿中记录cd光谱,扫描范围设置为200-600nm,扫描速率为100nm·min-1,狭缝宽度为1nm。实施例3~4制得的尾接有机导向分子的芳基钌配合物1,2与dna的作用如图1~2所示。
结果表明:随着[配合物]/[ct-dna]摩尔比的增加,ct-dna的圆二正负信号强度均逐渐减弱,其中,正峰强度减弱表明配合物与dna碱基对发生堆积作用,负峰强度减弱表明配合物通过插入碱基对模式使双螺旋dna发生部分解旋。这说明配合物1,2可能通过水解配位以及嵌入插入模式改变dna的二级结构。
方法二:琼脂糖凝胶电泳法。通过凝胶电泳研究配合物1,2与超螺旋质粒dnapbr322的相互作用。在50mm的tris-hcl(ph=7.4)缓冲溶液中配制一系列不同摩尔比的(ri=[配合物]/[ct-dna],ri值分别为0,1,2,3,4,5)样品,其中,ct-dna碱基对浓度设定为10μm,于37℃恒温箱中孵化24小时后,向内加入终止液(0.05%的溴酚蓝,50%的甘油和2mm的na2h2edta),将配制好的样品点样于提前铺制好的含有0.8%琼脂糖凝胶的孔槽中,在tae(40mmtris-acetate,1mmedta,ph=8.3)溶液中电泳,电压设为70mv,时间为120min,结束电泳后,使用10mg/μl的溴化乙锭染色20min,染色后的凝胶置于bio-rad分子成像geldocxr系统的紫外灯下成像,获得所需电泳图像,如图3~4所示。
结果表明:闭环质粒dna与药物结合后会改变其超螺旋密度,从而改变其在凝胶电泳中的迁移速率。随着配合物1,2浓度的增加,超螺旋环状dna带逐渐减少,而点样孔处的dna的量逐渐增加,这说明配合物可以诱导dna发生聚合,由于其巨大的分子量与复杂的结构影响dna在电泳中的迁移率从而无法发生迁移作用。配合物2与pbr322的相互作用结果显示,当r的值为3.0时,dna开始发生聚合现象,当r的值为5.0时,dna则发生完全聚合。配合物1与pbr322的作用结果与配合物2相似,不过配合物1对dna的聚合作用更加明显,当r的值为3.0时,超螺旋环状dna带消失,dna发生了完全聚合现象。以上结果表明,配合物1,2均能诱导dna发生聚合现象,但配合物1的作用更为明显,这可能是由于咪唑的亲水性强于联吡啶的亲水性。
本发明尾接有机导向分子的芳基钌配合物2对诱发细胞内活性氧生成的应用:
方法1:流式细胞仪监测活细胞内的ros。将50μm的配合物2加入到事先培养好的hela细胞中,于37℃,5%co2细胞培养箱中孵化1h,然后向内加入10μmdcfhda继续避光孵化30min。孵化结束后使用离心的方法获取细胞,并使用无血清的培养基洗涤细胞三次,除去未进入细胞内的dcfhd。最后上流式细胞仪(flowjo7.6software(treestar,or,usa))检测细胞内活性氧。激发光波长设为488nm,发射光波长设为530±30nm。
方法2:共聚焦显微镜监测细胞内的ros。将50μm的配合物2加入到事先培养好的hela细胞中,于37℃,5%co2细胞培养箱中孵化2h,然后向内加入10μmdcfhda继续避光孵化30min。孵化结束后使用无血清的培养基洗涤细胞三次,除去未进入细胞内的dcfhd。随后立刻使用共聚焦显微镜观察细胞荧光强度从而监测细胞内活性氧。激发光波长设为488nm,发射光波长设为530±30nm。
结果表明:配合物2明显可有效地诱发细胞内活性氧含量升高,共聚焦显微镜下加了药的荧光强度是未加药的2倍左右,这说明该类含有甘草次酸有机导向分子的芳基钌配合物可能通过诱发细胞内活性氧生成从而导致癌细胞发生凋亡。
本发明的桥联配体具有唯一能和氨基反应的官能团,同时,在配合物的合成过程中只存在唯一的配位点,这为后续合成分离带来方便,易于提高反应产物的产率;制得的具有有机导向分子的芳基钌配合物可用于后续分析研究具有不同结构的配合物与抗肿瘤/抗菌选择性之间的构效关系。在对本发明芳基钌配合物进一步抗癌机制的研究中发现,本发明芳基钌配合物能改变dna的二级结构并使dna发生聚合,同时还能诱发细胞内活性氧生成从而诱导癌细胞发生凋亡,从而为其作为制备抗肿瘤药物的发展提供了较好的参考价值。
- 还没有人留言评论。精彩留言会获得点赞!