遗传改造谷氨酸棒杆菌中DHDPR提高赖氨酸产量的方法与流程
本发明属于基因工程和酶工程领域,具体涉及遗传改造谷氨酸棒杆菌中dhdpr提高赖氨酸产量的方法。
技术背景
l-赖氨酸是人类和动物所必需的、自身不能合成的氨基酸,属于八大必需氨基酸之一。由于l-赖氨酸具有多种生理功能,如平衡氨基酸组成、调节体内代谢平衡、提高机体对谷类蛋白质的吸收及利用率和促进机体生长发育等,因而被广泛应用于饲料工业、医药工业及食品工业中。工业上生产l-赖氨酸的方法主要有三种:蛋白水解法、化学合成法和微生物发酵法,其中微生物发酵法具有生产成本低、生产强度高、高特异性和对环境污染小等优点而成为当今工业生产l-赖氨酸应用最广泛的方法。因此,选育具有自主知识产权的高l-赖氨酸合成、低副产物积累的生产菌株,开发具有自主知识产权的高纯度低色级的l-赖氨酸生产新技术、新工艺,对拓宽产品应用范围和利润空间,实现企业可持续发展具有重要意义。
用于生产l-赖氨酸的微生物包括多种属,其中国内外用于工业化生产l-赖氨酸的菌株多为谷氨酸棒杆菌(corynebacteriumglutamicum)及其亚种和大肠杆菌(escherichiacoli)的改造型菌株。l-赖氨酸生物合成不同于l-谷氨酸合成,c.glutamicum生物合成途径中合成1moll-赖氨酸需要消耗4molnadph,而形成1moll-谷氨酸只需要1molnadph。因此,为了提高c.glutamicum生物合成途径中l-赖氨酸的积累,提高c.glutamicum代谢途径中nadph量或降低l-赖氨酸合成途径中nadph需求量是非常重要的策略。
二氢吡啶二羧酸还原酶(dihydrodipicolinatereductase,dhdpr)是细菌和高等植物生物合成二氨基庚二酸和l-赖氨酸过程中第二个关键酶,催化二氢吡啶二羧酸的nad(p)h-依赖的还原性反应,生成六氢吡啶二羧酸。该酶在细胞壁形成中起到关键性作用。有活性的dhdpr一般以四分体的形式存在,单个亚基分子量大小约为30kda,每个亚基由两个区域组成,即c-端底物(即二氢吡啶二羧酸)或抑制子结合域和n-端二核苷酸结合域(即辅因子结合域),两个功能域由一个可变的环连接。n-端二核苷酸结合域由位于中心的7个平行β折叠和位于外围的4个α螺旋组成,辅因子结合位点位于β折叠的c-端边缘,而c-端底物或抑制子结合域由4个β折叠和2个α螺旋组成。dhdpr既以nadh又以nadph作为辅助因子,不同细菌的dhdpr对不同辅因子的亲和力也不同,如e.coli更青睐nadh,而c.glutamicum中dhdpr主要以nadph作为辅因子参与l-赖氨酸的合成。和其他已发现的生物体中的dhdpr一样,c.glutamicum中dhdpr由基因dapb编码,其酶活不受合成途径中终产物的调节作用,但受2,6-吡啶二羧酸(2,6-pdc)的抑制。前人研究结果表明,通过定点改变nadph-依赖型酶可以转变其对不同辅因子的亲和力。因此,通过定点突变c.glutamicum中dhdpr编码基因,改变dhdpr对nadph的亲和力,从而可以增加l-赖氨酸产量。
本实验室通过传统诱变方法,筛选得到的一株产l-赖氨酸突变菌株谷氨酸棒杆菌jl-6(c.glutamicumjl-6),该菌经摇瓶发酵产l-赖氨酸14.5g/l。然而,对该菌株l-赖氨酸发酵过程中胞内氧化还原辅因子测定时发现,胞内nadph含量依旧不足,从而限制了l-赖氨酸的进一步增加。另外,该菌株中dhdpr编码基因dapb也为发生错义突变。
基于研究室前期研究基础,本发明首先分析比较了不同微生物中dhdpr蛋白结构,找到了潜在的调节dhdpr结合不同辅因子的氨基酸残基:第11位lys残基和第13位arg残基。利用基因工程方法,定点突变dhdpr编码基因dapb,使dhdpr氨基酸序列的第11位lys残基和第13位arg残基突变成ala残基,获得重组菌株jl-6dapba31g,a32c(即lys11ala突变株,k11a)和jl-6dapbc37g,g38c(即arg13ala突变株,r13a)。这些重组菌株提高了dhdpr对nadh的亲和力而降低了对nadph的亲和力,解除了c.glutamicumjl-6发酵生产l-赖氨酸过程中nadph供应不足的缺点,提高了菌株积累l-赖氨酸的能力。
技术实现要素:
本发明的目的在于:定点突变c.glutamicumjl-6l-赖氨酸合成途径关键酶dhdpr编码基因dapb,改变dhdpr蛋白结构中辅因子结合域的结构,获得具有不同辅因子亲和力的重组菌株jl-6dapba31g,a32c和jl-6dapbc37g,g38c。这些重组菌株提高了dhdpr对nadh的亲和力而降低了对nadph的亲和力,解除了c.glutamicumjl-6发酵生产l-赖氨酸过程中nadph供应不足的缺点,提高了菌株积累l-赖氨酸的能力。该发明为成功改造l-赖氨酸合成途径中关键酶的蛋白质结构,改变其辅因子亲和力,解除合成过程中nadph供应不足提供了一条崭新的思路。
本发明的技术方案:以基因工程为手段,通过改变二氢吡啶二羧酸还原酶的氧化还原辅因子亲和力,缓解l-赖氨酸合成过程中nadp(h/+)供应不足,获得高产l-赖氨酸的重组菌株。
(1)改变了二氢吡啶二羧酸还原酶的氧化还原辅因子亲和力,可高效积累l-赖氨酸的重组谷氨酸棒杆菌,其分类命名分别为c.glutamicumjl-6dapba31g,a32c和c.glutamicumjl-6dapbc37g,g38c。
(2)所述重组菌的构建方法,首先利用在线软件比较分析不同微生物来源的dhdpr,确定潜在的调节dhdpr结合不同辅因子的氨基酸残基:第11位lys残基和第13位arg残基;利用定点突变试剂盒分别突变dhdpr编码基因dapb,随后利用携带抗生素抗性与条件致死型标记sacb双标记的新型整合型载体pk18mobsacb,通过两次同源重组替换c.glutamicumjl-6中自身基因dapb,分别获得重组菌株c.glutamicumjl-6dapba31g,a32c和c.glutamicumjl-6dapbc37g,g38c。
①潜在的调节dhdpr结合不同辅因子的氨基酸残基的确定
来源于e.coli和结核杆菌(mycobacteriumtuberculosis)的dhdpr更青睐利用nadh作为辅因子,而来源于c.glutamicum和海栖热袍菌(thermotogamaritima)的dhdpr更青睐利用nadph作为辅因子。为此通过采用在线软件clustalw和espript3.0比较分析它们dhdpr结构中的共同点与不同点,从而找出潜在的调节dhdpr结合不同辅因子的氨基酸残基:第11位lys残基和第13位arg残基。最后,采用pymol软件分析不同突变体的dhdpr蛋白结构发现,分别将第11位lys残基和第13位arg残基突变成ala,会改变dhdpr中与nadph的2’-磷酸残基的结构,从而影响dhdpr与nadph的亲和性。因此,选择dhdpr氨基酸序列中第11位lys残基和第13位arg残基作为后期实验改造位点。
②c.glutamicum中dhdpr编码基因dapb的体外定点突变
根据genbank中c.glutamicumatcc13032dapb基因序列设计dapb基因上下游以及定点突变引物,引物序列如表1:
表1.pcr扩增所需引物序列(下划线为酶切位点)
以c.glutamicumjl-6基因组为模板,以dapb-f/dapb-r为引物pcr,获得dapb片段,其全长747bp。将上述dapb片段与t载体(pucm-t)连接,转化大肠杆菌,提取重组质粒pucm-t/dapb。以质粒pucm-t/dapb为模板,分别以突变引物mcb1-f/mcb1-r和mcb2-f/mcb2-r为引物进行pcr反应,用dna片段纯化试剂盒纯化后用限制性内切酶dpni酶切(dpni只识别甲基化的dna,而新合成的dna不被甲基化),随后将酶切产物纯化后转化到e.colijm106感受态细胞中,并涂布于氨苄抗性平板中筛选突变菌株,最后挑起在氨苄抗性平板生长良好的菌株培养,提取质粒并测序鉴定,获得目的重组质粒pucm-t/dapba31g,a32c和pucm-t/dapbc37g,g38c。
③dhdpr突变体dhdprk11a和dhdprr13a的动力学测定
采用相应的限制性内切酶分别酶切重组质粒pucm-t/dapba31g,a32c和pucm-t/dapbc37g,g38c。随后采用胶回收试剂盒回收dapba31g,a32c和dapbc37g,g38c片段。将dapba31g,a32c和dapbc37g,g38c片段分别与经相同限制性内切酶酶切后的带有his标签的表达质粒pet28a相连构建重组质粒pet28a/dapba31g,a32c和pet28a/dapbc37g,g38c。分别将验证正确的质粒pet28a/dapba31g,a32c和pet28a/dapbc37g,g38c转化到e.colibl21感受态细胞中,经lb+km固体培养基筛选,获得重组转化子。将目的转化子转接至lb液体培养基中iptg诱导表达,并利用蛋白质纯化仪纯化目的蛋白。最后考查突变体dhdprk11a、dhdprr13a和野生型dhdpr对nadh和nadph的动力学参数。
④重组菌c.glutamicumjl-6dapba31g,a32c和c.glutamicumjl-6dapbc37g,g38c的获得
采用相应的限制性内切酶分别酶切重组质粒pucm-t/dapba31g,a32c和pucm-t/dapbc37g,g38c。随后采用胶回收试剂盒回收dapba31g,a32c和dapbc37g,g38c片段。将dapba31g,a32c和dapbc37g,g38c片段分别与经相同限制性内切酶酶切后的线性化pk18mobsacb整合型载体相连构建重组质粒pk18mobsacb/dapba31g,a32c和pk18mobsacb/dapbc37g,g38c。分别将验证正确的质粒pk18mobsacb/dapba31g,a32c和pk18mobsacb/dapbc37g,g38c电击转化c.glutamicumjl-6,经lbhis+km固体培养基筛选,获得第一次同源重组转化子。再分别将目标转化子在含蔗糖的培养基中进行胁迫二次重组筛选,最终在lbg平板上进行划线分离并挑取多个转化子。对发生第二次同源重组的菌株进行回复野生型/基因突变型的鉴定。提取转化子染色体,以目标基因dapb的上下游引物进行pcr并对pcr产物进行测序鉴定。鉴定正确的转化子命名为c.glutamicumjl-6dapba31g,a32c和c.glutamicumjl-6dapbc37g,g38c。
(3)重组菌c.glutamicumjl-6dapba31g,a32c和c.glutamicumjl-6dapbc37g,g38c发酵产l-赖氨酸
挑取单菌落接种至液体种子培养基,30℃,100rmin-1往复式摇床培养12h。以10%接种量将种子培养液转接至发酵培养基,30℃,100rmin-1往复式摇床培养40h。实时检测发酵过程中l-赖氨酸、葡萄糖残留量以及菌体量等参数。
(4)底物与产物的定性与定量分析以及菌体生长情况的监测
发酵液中葡萄糖和l-赖氨酸的实时检测通过sba-40b生物传感分析仪测定。菌液浓度测定:吸取样品菌液,用蒸馏水稀释一定倍数,以蒸馏水作为空白对照,采用分光光度计于1cm光程测定od600nm。
(5)副产物的定性与定量分析
发酵液中副产物主要考查有机酸和氨基酸。对于有机酸的测定,采用高效液相色谱仪(hlpc)测定;对于氨基酸的测定,采用氨基酸测定仪测定。采用高效液相色谱仪或氨基酸测定仪分别测定标准样品和转化液各自的出峰时间和峰面积,即可以对转化液中待测物质进行定性与定量检测。
本发明的有益效果:通过定点突变c.glutamicumjl-6l-赖氨酸合成途径关键酶dhdpr编码基因dapb,改变dhdpr蛋白结构中辅因子结合域的结构,获得了更青睐利用nadh作为辅因子的重组菌株jl-6dapba31g,a32c和jl-6dapbc37g,g38c,从而解除了l-赖氨酸发酵生产过程中nadph供应不足的缺点,提高了菌株积累l-赖氨酸的能力。
附图说明
图1不同微生物来源的dhdpr的氨基酸序列比对;
缩写说明:cgdhdpr-来源于c.glutamicum的dhdpr;mtdhdpr-来源于m.tuberculosis的dhdpr;ecdhdpr–来源于e.coli的dhdpr;tmdhdpr–来源于t.maritima的dhdpr。
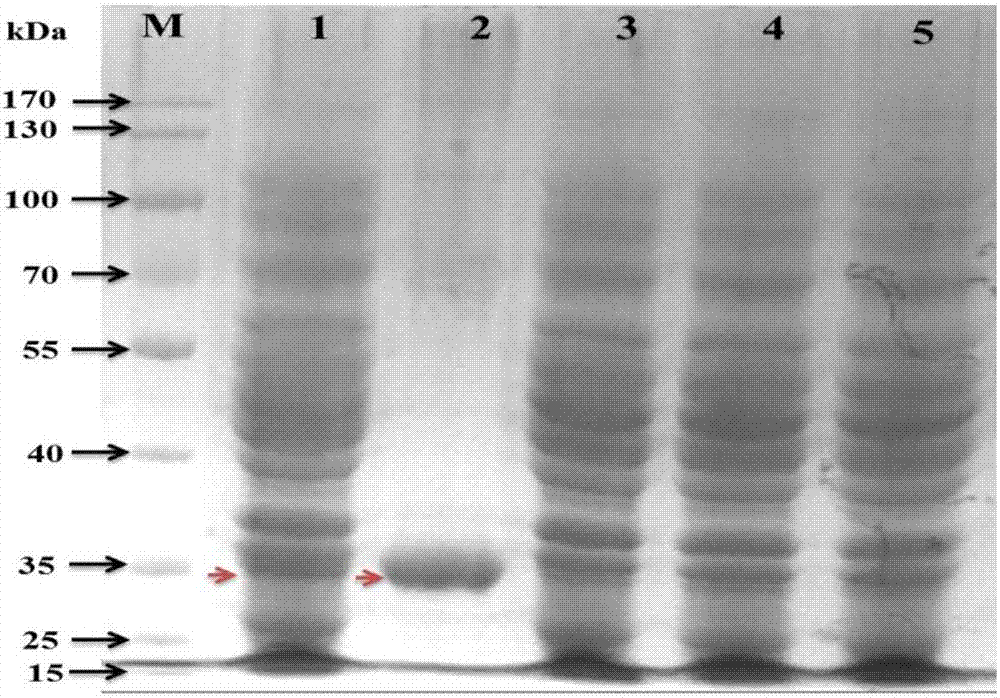
图2dhdpr在e.colibl21中的表达;
泳道说明:m泳道为蛋白质分子量标准marker;1号泳道为e.colibl21(pet28a/dapb)细胞抽提液加iptg诱导;2号泳道为e.colibl21(pet28a/dapb)细胞抽提液纯化蛋白;3号泳道为e.colibl21(pet28a/dapb)细胞抽提液未诱导;4号泳道为e.colibl21(pet28a)细胞抽提液;5号泳道为e.colibl21细胞抽提液。
图3不同dhdpr突变株dhdpr酶活力测定;
图4不同dhdpr突变株发酵生产l-赖氨酸过程曲线;
编号说明:a-l-赖氨酸变化曲线;b-葡萄糖变化曲线;c-菌体生长曲线。
图5不同dhdpr突变株发酵液副产物测定结果;
编号说明:a-有机酸积累量;b-氨基酸积累量
具体实施方法
实施例1:c.glutamicum中dhdpr编码基因dapb的体外定点突变
以c.glutamicumjl-6基因组为模板,以dapb-f/dapb-r为引物,通过pcr反应扩增747bp的dapb片段,在pcr产物的5’和3’端分别引入ecori和hindiii限制性酶切位点。
将上述dapb片段与t载体(pucm-t)连接,转化大肠杆菌,提取重组质粒pucm-t/dapb。以质粒pucm-t/dapb为模板,分别以突变引物mcb1-f/mcb1-r和mcb2-f/mcb2-r为引物进行pcr反应,用dna片段纯化试剂盒纯化后用限制性内切酶dpni酶切(dpni只识别甲基化的dna,而新合成的dna不被甲基化),随后将酶切产物纯化后转化到e.colijm106感受态细胞中,并涂布于氨苄抗性平板中筛选突变菌株,最后挑起在氨苄抗性平板生长良好的菌株培养,提取质粒并测序鉴定,获得目的重组质粒pucm-t/dapba31g,a32c和pucm-t/dapbc37g,g38c。
pcr反应体系(50μl):extaq酶25μl,dna模板100μg,正向引物(20μmol·l-1)1μl,反向引物(20μmol·l-1)1μl,加ddh2o至50μl;pcr反应条件:首先94℃预变性5min,然后94℃变性30s,退火30s,72℃延伸,计35个循环,最后72℃再延伸10min并4℃保存。反应中的退火温度和延伸时间如表1所示。
实施例2:dhdpr突变体在e.colibl21中的表达及纯化
根据表1指出,采用限制性内切酶ecori和hindiii分别酶切重组质粒pucm-t/dapba31g,a32c和pucm-t/dapbc37g,g38c。随后采用胶回收试剂盒回收dapba31g,a32c和dapbc37g,g38c片段。将dapba31g,a32c和dapbc37g,g38c片段分别与经相同限制性内切酶酶切后的pet28a相连构建重组质粒pet28a/dapba31g,a32c和pet28a/dapbc37g,g38c;将重组质粒pet28a/dapba31g,a32c和pet28a/dapbc37g,g38c转入e.colibl21,在卡那霉素抗性lb平板上挑取阳性转化子至液体lb,iptg诱导后收集菌体经超声波破碎后,上清液sds-page,检测到一条分子量约30kda的特异性条带(图2),与报道的目标蛋白大小一致,而对照菌株e.colibl21(pet28a)均无此条带。说明重组质粒pet28a/dapba31g,a32c和pet28a/dapbc37g,g38c在大肠杆菌中正确表达。随后,采用蛋白质纯化仪,根据表达质粒pet28a上的his标签纯化细胞破碎液中dhdpr蛋白(图2)。
实施例3:dhdpr突变体动力学参数的测定
分别取上述纯化的突变体dhdprk11a、dhdprr13a和野生型dhdpr加入dhdpr酶活测定反应体系中,实时监测340nm处吸光度变化,并根据吸光度变化计算处km和kcat(表2)。
反应体系:100mmoll-1mops缓冲液(ph7.2)、50μmoll-1二氢吡啶二羧酸、0.01~0.04mmoll-1nadph或0.004~0.032mmoll-1nadh;反应温度:30℃;反应时间:≥300s。
表2.野生型dhdpr及其突变体动力学参数
实施例4:重组菌c.glutamicumjl-6dapba31g,a32c和c.glutamicumjl-6dapbc37g,g38c的获得
根据表1指出,采用限制性内切酶ecori和hindiii分别酶切重组质粒pucm-t/dapba31g,a32c和pucm-t/dapbc37g,g38c。随后采用胶回收试剂盒回收dapba31g,a32c和dapbc37g,g38c片段。将dapba31g,a32c和dapbc37g,g38c片段分别与经相同限制性内切酶酶切后的线性化pk18mobsacb整合型载体相连构建重组质粒pk18mobsacb/dapba31g,a32c和pk18mobsacb/dapbc37g,g38c。分别将验证正确的重组质粒pk18mobsacb/dapba31g,a32c和pk18mobsacb/dapbc37g,g38c电击转化c.glutamicumjl-6,随后将转化液涂布于含有25μgml-1卡那霉素的lbhis固体培养基筛选,挑选lbhis+km平板生长良好的菌株,获得第一次同源重组转化子。再分别将目标转化子接种于lbg液体培养基中培养24h后,倍比稀释后涂布于含100gl-1蔗糖的lbg固体培养基中进行胁迫二次重组筛选,最终在lbg平板上进行划线分离并挑取多个转化子。提取转化子基因组,以目标基因dapb的上下游引物进行pcr并对pcr产物进行测序鉴定,确定c.glutamicumjl-6dapb基因发生定点突变。鉴定正确的转化子命名为c.glutamicumjl-6dapba31g,a32c和c.glutamicumjl-6dapbc37g,g38c。
实施例5:重组菌c.glutamicumjl-6dapba31g,a32c和c.glutamicumjl-6dapbc37g,g38c的dhdpr酶活力测定
取冻管保藏的菌种接种含有0.25gl-1的l-蛋氨酸和40gl-1葡萄糖的cgxii培养基(即cgxiimg培养基)中,30℃振荡培养过夜,并于10000rmin-1离心收集菌体。随后,将菌体悬浮于mops缓冲液中并于超声波破碎法制备粗酶液。粗酶液采用比色法进行酶活测定(a320nm)。酶反应体系:100mmoll-1mops缓冲液(ph7.2)、50μmoll-1二氢吡啶二羧酸、0.162mmoll-1nadph或0.162mmoll-1nadh;反应温度:30℃;反应时间:≥300s。一个酶活力单位(u)定义为在测定条件下每分钟氧化1μmolnadph或nadh所需的酶量。经测定,重组菌dhdpr酶活力都要高于对照菌株,结果如图3所示。
实施例6:重组菌c.glutamicumjl-6dapba31g,a32c和c.glutamicumjl-6dapbc37g,g38c胞内辅因子的测定
取谷氨酸棒杆菌单菌落接种于cgxiimg液体培养基中,30℃、100r·min-1摇床培养约10h,4℃,6000r·min-1离心收集菌体,并洗涤菌体三次,去除残留的细胞外代谢物。随后,以酸性抽提液(0.5mol·l-1hcl)提取氧化型吡啶核苷酸(nad+和nadp+),以碱性抽提液(0.5mol·l-1naoh)提取还原型吡啶核苷酸(nadh和nadph)。随后,借助从biovision公司购置的定量分析试剂盒,利用酶循环法测定nad(p)+和nad(p)h的浓度并计算nadh/nad+和nadph/nadp+,其中以nad/nadhquantificationcolorimeterickit特异性检测nad+和nadh,以nadp/nadphquantificationcolorimeterickit特异性检测nadp+和nadph,具体步骤参照试剂盒说明书的方法进行,结果如表3所示。
实施例7:c.glutamicumjl-6dapba31g,a32c和c.glutamicumjl-6dapbc37g,g38c发酵产l-赖氨酸
培养基:①种子培养基(g·l-1):葡萄糖5,蛋白胨10,酵母膏5,nacl10,ph7.0,121℃灭菌20min;②发酵培养基(即cgxiimg;g·l-1):葡萄糖40,(nh4)2so420,尿素5,kh2po41,k2hpo4·3h2o1,mgso4·7h2o0.25,3-(n-吗啡啉)-丙磺酸42,l-蛋氨酸0.25,cacl20.010,feso4·7h2o0.01,mnso4·h2o0.01,znso4·7h2o0.01,cuso4·5h2o0.0002,nicl2·6h2o0.00002,生物素0.0002,原儿茶酸0.00003,115℃灭菌10min。发酵过程中通过添加10g·l-1caco3调节ph。
将上述经验证的重组c.glutamicumjl-6dapba31g,a32c和c.glutamicumjl-6dapbc37g,g38c分别进行摇瓶发酵实验,从新鲜活化的斜面培养基中挑取一满环c.glutamicum(对照菌及重组菌)于种子培养基中(25ml/250ml),30℃,1000rmin-1往复式摇床培养12h;以10%接种量,接种于发酵培养基中,30℃,1000rmin-1往复式摇床培养40h,分时间区段测定l-赖氨酸、葡萄糖及生物量,结果与对照菌株c.glutamicumjl-6相比,结果如图4所示。
此外,分别采用高效液相色谱仪和氨基酸分析仪测定重组菌c.glutamicumjl-6dapba31g,a32c和c.glutamicumjl-6dapbc37g,g38c以及对照菌c.glutamicumjl-6中副产物积累情况(包括有机酸和氨基酸),结果如图5所示。
以上所述方法不仅适用于利用谷氨酸棒杆菌发酵生产l-赖氨酸,对其它任何利用微生物发酵生产nadph-依赖型产物都适用,因此这些内容都属于本发明的保护范围。
序列表
<110>江南大学
<120>遗传改造谷氨酸棒杆菌中dhdpr提高赖氨酸产量的方法
<160>3
<170>siposequencelisting1.0
<210>1
<211>747
<212>dna
<213>corynebacteriumglutamicum
<400>1
atgggaatcaaggttggcgttctcggagccaaaggccgtgttggtcaaactattgtggca60
gcagtcaatgagtccgacgatctggagcttgttgcagagatcggcgtcgacgatgatttg120
agccttctggtagacaacggcgctgaagttgtcgttgacttcaccactcctaacgctgtg180
atgggcaacctggagttctgcatcaacaacggcatttctgcggttgttggaaccacgggc240
ttcgatgatgctcgtttggagcaggttcgcgactggcttgaaggaaaagacaatgtcggt300
gttctgatcgcacctaactttgctatctctgcggtgttgaccatggtcttttccaagcag360
gctgcccgcttcttcgaatcagctgaagttattgagctgcaccaccccaacaagctggat420
gcaccttcaggcaccgcgatccacactgctcagggcattgctgcggcacgcaaagaagca480
ggcatggacgcacagccagatgcgaccgagcaggcacttgagggttcccgtggcgcaagc540
gtagatggaatcccggttcatgcagtccgcatgtccggcatggttgctcacgagcaagtt600
atctttggcacccagggtcagaccttgaccatcaagcaggactcctatgatcgcaactca660
tttgcaccaggtgtcttggtgggtgtgcgcaacattgcacagcacccaggcctagtcgta720
ggacttgagcattacctaggcctgtaa747
<210>2
<211>747
<212>dna
<213>corynebacteriumglutamicum
<400>2
atgggaatcaaggttggcgttctcggagccgcaggccgtgttggtcaaactattgtggca60
gcagtcaatgagtccgacgatctggagcttgttgcagagatcggcgtcgacgatgatttg120
agccttctggtagacaacggcgctgaagttgtcgttgacttcaccactcctaacgctgtg180
atgggcaacctggagttctgcatcaacaacggcatttctgcggttgttggaaccacgggc240
ttcgatgatgctcgtttggagcaggttcgcgactggcttgaaggaaaagacaatgtcggt300
gttctgatcgcacctaactttgctatctctgcggtgttgaccatggtcttttccaagcag360
gctgcccgcttcttcgaatcagctgaagttattgagctgcaccaccccaacaagctggat420
gcaccttcaggcaccgcgatccacactgctcagggcattgctgcggcacgcaaagaagca480
ggcatggacgcacagccagatgcgaccgagcaggcacttgagggttcccgtggcgcaagc540
gtagatggaatcccggttcatgcagtccgcatgtccggcatggttgctcacgagcaagtt600
atctttggcacccagggtcagaccttgaccatcaagcaggactcctatgatcgcaactca660
tttgcaccaggtgtcttggtgggtgtgcgcaacattgcacagcacccaggcctagtcgta720
ggacttgagcattacctaggcctgtaa747
<210>3
<211>747
<212>dna
<213>corynebacteriumglutamicum
<400>3
atgggaatcaaggttggcgttctcggagccaaaggcgctgttggtcaaactattgtggca60
gcagtcaatgagtccgacgatctggagcttgttgcagagatcggcgtcgacgatgatttg120
agccttctggtagacaacggcgctgaagttgtcgttgacttcaccactcctaacgctgtg180
atgggcaacctggagttctgcatcaacaacggcatttctgcggttgttggaaccacgggc240
ttcgatgatgctcgtttggagcaggttcgcgactggcttgaaggaaaagacaatgtcggt300
gttctgatcgcacctaactttgctatctctgcggtgttgaccatggtcttttccaagcag360
gctgcccgcttcttcgaatcagctgaagttattgagctgcaccaccccaacaagctggat420
gcaccttcaggcaccgcgatccacactgctcagggcattgctgcggcacgcaaagaagca480
ggcatggacgcacagccagatgcgaccgagcaggcacttgagggttcccgtggcgcaagc540
gtagatggaatcccggttcatgcagtccgcatgtccggcatggttgctcacgagcaagtt600
atctttggcacccagggtcagaccttgaccatcaagcaggactcctatgatcgcaactca660
tttgcaccaggtgtcttggtgggtgtgcgcaacattgcacagcacccaggcctagtcgta720
ggacttgagcattacctaggcctgtaa747
- 还没有人留言评论。精彩留言会获得点赞!