一种用于微量样本个体识别及亲子鉴定的方法与流程
本发明涉及法医学鉴定领域,尤其涉及一种用于微量样本个体识别及亲子鉴定的方法。
背景技术:
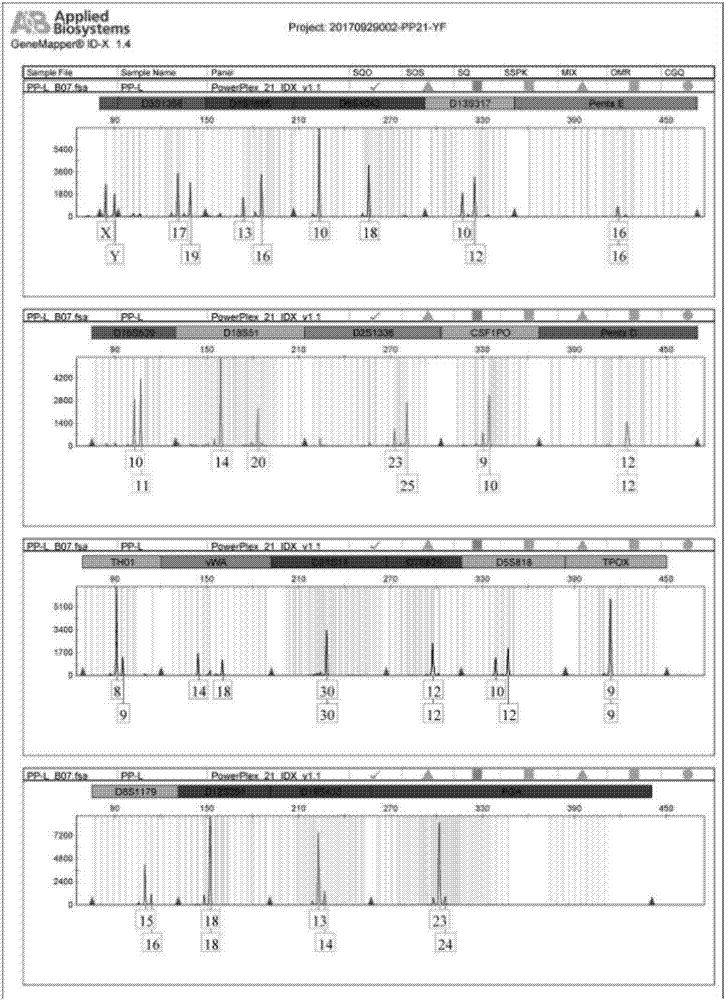
:基因是dna分子上携带有遗传信息的功能片断,是生物传递遗传信息的物质。dna的使用日益广泛,特别是在刑事司法实践中,不断发展完善并在法庭科学领域发挥了巨大作用。法庭科学dna数据库现已成为主流dna数据库中涉及人员样本最广、也最为公众熟知的数据库。自从pcr技术应用到生物物证的检验,尤其是应用到str基因座进行分析后,dna提取便成为法医dna多态性分析的关键环节,而dna样品质量的好坏将直接关系到后续实验的成败。伴随高通量测序技术的快速发展,单核苷酸多态性(snp)作为第三代遗传标记,越来越多的成为个体及亲子鉴定最新的检测手段。相对于str,snp在染色体中的分布更为广泛,数量更多,检测方法也更方便可靠。但这并不意味着二代测序技术能够即刻常规应用于法庭科学:第一,目前二代测序的成本较高,不能被法医学应用广泛接受;第二,二代测序平台都不可避免地存在一定比例的错误率,技术解决方案还未完全成熟完整,达不到dna数据库引入要求;最后,二代测序解读规则尚未制定,不能应用于全国实验室数据共享范围。因此,对已有str数据库的充分应用成为目前法医鉴定最经济实惠的检测方案。在实际检案中,越来越多的案件由于案件本身性质、作案过程或犯罪分子的反侦察意识增强等因素的作用,常规法医鉴定中难免会遇到微量样本,由于采样困难、样本降解等原因,使得dna抽提困难从而达不到检测输入量而影响str扩增鉴定。为了解决这一问题,本发明涉及一种针对微量样本的个体识别及亲子鉴定的方法,可将单细胞(pg,皮克)级的样本总量扩增至常规str鉴定需要的纳克级(约150-150000个细胞dna量),整个流程无需纯化及转管即可进行str鉴定,同时可以防止纯化及转管带来的样本损失或污染,从而使得样本损失更小,污染更少。如上所述,微量样本或单细胞dna的数量极其微小,对其分析之前必须进行dna的扩增。而现有技术中,以pcr(聚合酶链式反应)为基础的全基因组扩增技术,在pcr反应过程中,dna片段的差异会影响聚合酶的扩增效率甚至导致酶滑链或者从模板上脱离,不能完整地覆盖基因组,并且会向序列中引入很多错误和非特异性扩增产物,存在扩增偏倚,在起始样本非常稀少时,使扩增质量大大降低,并且一次只能通过特定引物对单个基因位点进行检查,无法对同一样品进行多位点扩增和检测,无法进行单细胞全基因的分析。而新型的基于mda(多重置换扩增)的扩增方法,在现有方法下也存在扩增偏倚量大、扩增质量不好、假阳性等现象。因此为解决单细胞扩增中扩增偏倚量大、扩增质量不好、假阳性等问题,本发明同时创造性地设计并采用特殊修饰引物,避免了非特异性扩增产生,同时利用平行样本分析方案,解决可能存在的等位基因缺失问题。简而言之,本发明提供了一种更好的高效、简单、实用、精准、污染小、损失少、成本低、方法重复性好、非常适合微量样本的应用的个体识别及亲子鉴定的方法,为法医及日常鉴定拓展了样本应用范围,同时提高了检测的精密度。技术实现要素:鉴于现有技术的不足,本专利旨在提供一种更佳的单细胞扩增的改进方法,旨在利用改进的单细胞扩增方法对微量样本进行扩增,通过对扩增产物进行str检测从而获得样本str分型结果。本发明创造性地将一种改进的较佳单细胞扩增方法对微量人类样本进行扩增,对扩增产物进行str检测即可获得样本str分型结果。流程为样本裂解(从样本中释放基因组dna)-样本扩增(将得到的皮克级别dna通过扩增方式富集为纳克甚至微克级)-str鉴定(基因座位点扩增及毛细管电泳鉴定),整个流程无需转管及纯化,2-3小时即可完成。比对参考序列即可得知样本与参考序列之间的关系鉴定结果。不仅仅是创造性地将一种改进的最佳单细胞扩增方法首次应用到微量样本,更重要的是创造性地设计并采用特殊修饰引物,避免了非特异性扩增产生,同时利用平行样本分析方案,解决可能存在的等位基因缺失问题。为实现上述目的及其他相关目的,本发明提供了一种用于微量样本个体识别及亲子鉴定的方法,包括如下步骤:(1)样本裂解:向样本中加入细胞裂解液,使微量样本中的dna释放,获取基因组dna;进一步地,所述细胞裂解液ph=14;进一步地,所述细胞裂解液包括但不限于以下一种或几种:0.2mnaoh+1%sds、0.4mkoh+80mmdtt、400mmkoh+30mmedta+10mmdtt、300mmkoh+10mmedta等,最优的为300mmkoh+10mmedta;需要指出,可以是在进行步骤(2)之前,也可以采用由他人利用本领域所熟知的技术已经由微量或单细胞样本裂解获得或由多细胞样本抽提获得的样本基因组dna。进一步地向经过步骤(1)处理后获得的细胞裂解液产物中加入中和液,进行样本中和;进一步地,所述中和液ph=4;进一步地,所述中和液包括但不限于以下一种或几种:0.1m醋酸钠、0.1mmhcl、1mtris-hcl等,最优的为1mtris-hcl;(2)样本扩增:将经过步骤(1)处理后获得的产物中直接加入扩增缓冲液,使基因组dna进行样本扩增;进一步地,所述扩增缓冲液包括:dna聚合酶、bsa、随机引物、dntp、dna聚合酶、无核酸水;进一步地,所述dna聚合酶buffer稀释至1乘;所述bsa为0.1~1mg/ml,优选地为0.2mg/m;所述随机引物为1~50μm,优选地为30μm;所述dntp为0.2~1μm,优选地为0.6μm;所述dna聚合酶为0.2~2u,优选地为0.4u;所述无核酸水(补齐至总体系50μl)进一步地,所述dna聚合酶包括但不限于以下一种或几种:klenow片段(3′→5′exo-)、klenow片段、bstdna聚合酶、ventdna聚合酶(3′→5′exo-)、ventdna聚合酶、phi29dna聚合酶、deepventdna聚合酶(3′→5′exo-)、deepventdna聚合酶;进一步地,所述随机引物为具有特殊修饰的引物,包括但不限于以下任一种:3’端的两个碱基进行硫代修饰,碱基组成可以是n5引物(n=a/t/c/g)、d5引物(d=a/g/t)、h5引物(h=a/c/t)、r5引物(r=a/g)、w5引物(w=a/c)、m5引物(m=a/c)、n6引物(n=a/t/c/g)、d6引物(d=a/g/t)、h6引物(h=a/c/t)、r6引物(r=a/g)、w6引物(w=a/c)、m6引物(m=a/c)、n7引物(n=a/t/c/g)、d7引物(d=a/g/t)、h7引物(h=a/c/t)、r7引物(r=a/g)、w7引物(w=a/c)、m7引物(m=a/c)等任意一种,最优的为w5、w6、w7引物;进一步地,所述随机引物的浓度为10-50μm,最优的为30μm;进一步地,所述样本扩增为等温扩增,在pcr仪上进行如下程序:30℃孵育1-6h,65℃孵育3minutes,4℃保存。优选地30℃孵育时间为3h。(3)str鉴定:将经过步骤(2)处理获得的扩增产物进行str基因座位点扩增及毛细管电泳鉴定;进一步地,所述str基因座位点可采用多次平行样本位点进行总结分析,避免等位基因缺失,从而得到的str最终位点即可进行亲权关系计算;进一步地,所述步骤(3)也可使用双链dna染料定量经过步骤(2)获得扩增产物,使用str扩增试剂盒进行鉴定。此步骤中的str扩增试剂盒可以是以下任意一种,ab公司的identifilertm、direct、plus、sinofiler和promega公司的21system、国产试剂盒dna15plus、agcu17+1、goldeneyetm20a、阅微mr21id等。最优的为21system法医检测试剂盒。需要说明的是,样本进行实验操作时最好采用一式2~5的平行对照体系,较优地采用3次,可根据实验结果进行调整和补充重复实验。需要指出,上述方案可适用于珍贵样本、混合样本、降解样本的str分型鉴定。本申请提供了一种更佳的单细胞扩增的改进方法,旨在利用改进的单细胞扩增方法对微量样本进行扩增,通过对扩增产物进行str检测从而获得样本str分型结果。与现有技术相比,本发明所提供的技术具有以下优点:1.本发明的设计方案,可将单细胞(pg,皮克)级的样本总量扩增至常规str鉴定需要的纳克级(约150-150000个细胞dna量),直接通过裂解去除蛋白及细胞杂质,无需进行dna抽提以及纯化,即可进行str鉴定,步骤更简单、效果更精准,同时克服了现有技术中微量样本采样困难、样本易降解等原因导致的dna抽提困难从而达不到检测输入量而影响str扩增鉴定的技术难题。简而言之,本申请提供的设计方案步骤更简单、效果更精准、方法重复性好、成本较低、时间周期较短、应用更广泛。2.本发明的设计方案适用对象广泛,可用于珍贵样本、混合样本、降解样本的str分型鉴定,因此具有广泛的应用前景。3.本发明的设计方案中裂解及中和缓冲液成分适用于dna扩增酶,可直接对裂解得到的dna进行扩增,步骤更简单、更直接。4.本发明的设计方案整个流程无需纯化及转管,可以防止纯化及转管带来的样本损失或污染,因此本发明的设计方案样本损失更小,污染更少。5.本发明的提供了一种更佳的单细胞扩增的改进方法,采用等温扩增方式,使得本发明的设计方案保真性更强。6.本发明的设计方案创造性地设计并采用特殊修饰引物,降低引物自连导致的非特异性扩增效应,防止假阳性问题的产生。7.本发明的设计方案创造性地通过对dna的扩增可达到str检测试剂盒起始量,大大增强了str检测的应用范围,对微量基因组样本、获得的稀有细胞鉴定、单克隆细胞鉴定、个体识别、亲缘鉴定等具有良好广泛的应用价值。附图说明图1a是一个较佳实施例1的单细胞样本pp-b的str基因分型鉴定结果图图1b是一个较佳实施例1的单细胞样本pp-md的str基因分型鉴定结果图图1c是一个较佳实施例1的单细胞样本pp-l的str基因分型鉴定结果图图2a是一个较佳实施例2的单细胞样本dmd1的str基因分型鉴定结果图图2b是一个较佳实施例2的单细胞样本dmd2的str基因分型鉴定结果图图2c是一个较佳实施例2的单细胞样本fmd1的str基因分型鉴定结果图图2d是一个较佳实施例2的单细胞样本fmd2的str基因分型鉴定结果图图2e是一个较佳实施例2的单细胞扩增阴性对照样本n的str基因分型鉴定结果图具体实施方式下面结合具体实施例,进一步阐述本发明。应理解,实施方式只是为了举例说明本发明,而非以任何方式限制发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件或按照制造厂商所建议的条件。除非另行定义,文中所使用的所有专业与科学用语与本领域熟练人员所熟悉的意义相同。此外,任何与所记载内容相似或均等的方法及材料皆可应用于本发明方法中。文中所述的较佳实施方法与材料仅作示范之用。本发明技术方案1、样本收集将样本收集于1-6μl细胞收集液中。此步骤中的细胞收集液可以是无菌生理盐水、1*pbs、hanks'缓冲液、葡萄糖缓冲液、dulbecco's磷酸盐缓冲等细胞等渗溶液,最优的为1*pbs。此步骤中的细胞收集液体积范围为1-6μl,优选的为4μl.需要指出,样本收集可采用如上方法,也可以采用由他人利用本领域所熟知的技术已经采集好的微量样本。2、样本裂解向上述细胞收集液中加入1-6μl细胞裂解液,此时缓冲液体系总体积为7μl。此步骤中的细胞裂解液也可以是其他ph=14的缓冲液,如0.2mnaoh+1%sds、0.4mkoh+80mmdtt、400mmkoh+30mmedta+10mmdtt、300mmkoh+10mmedta等,最优的为300mmkoh+10mmedta。此步骤中的细胞裂解液体积范围为1-6μl,优选的为3μl。需要指出,样本裂解可采用如上方法,也可以采用由他人利用本领域所熟知的技术已经由微量或单细胞样本裂解获得或由多细胞样本抽提来获得样本基因组dna。3、样本中和向上述细胞裂解液产物加入3μl中和液,此时缓冲液体系总体积为10μl。此步骤中的中和液也可以是其他ph=4的缓冲液,如0.1m醋酸钠、0.1mmhcl、1mtris-hcl等,最优的为1mtris-hcl。4、样本扩增向上述样本中和液产物中直接加入扩增缓冲液(配制见下表1),此时缓冲液体系总体积为50μl。进行等温扩增,在pcr仪上进行如下程序:30℃孵育1-6h,65℃孵育3minutes,4℃保存。优选地30℃孵育时间为3h。表1试剂浓度dna聚合酶buffer稀释至1乘bsa0.2mg/ml随机引物10μmdntp0.6μmdna聚合酶0.4u/μl无核酸水补齐至总体系50μl此步骤中dna聚合酶可以是klenow片段(3′→5′exo-)、klenow片段、bstdna聚合酶、ventdna聚合酶(3′→5′exo-)、ventdna聚合酶、phi29dna聚合酶、deepventdna聚合酶(3′→5′exo-)、deepventdna聚合酶中的一种或几种混合物。相应的扩增缓冲液10*phi29buffer也可以是上述酶对应的任一种或多种buffer。此步骤中的随机引物为具有特殊修饰的引物,3’端的两个碱基进行硫代修饰,碱基组成可以是n5引物(n=a/t/c/g)、d5引物(d=a/g/t)、h5引物(h=a/c/t)、r5引物(r=a/g)、w5引物(w=a/c)、m5引物(m=a/c)、n6引物(n=a/t/c/g)、d6引物(d=a/g/t)、h6引物(h=a/c/t)、r6引物(r=a/g)、w6引物(w=a/c)、m6引物(m=a/c)、n7引物(n=a/t/c/g)、d7引物(d=a/g/t)、h7引物(h=a/c/t)、r7引物(r=a/g)、w7引物(w=a/c)、m7引物(m=a/c)等任意一种,最优的为w5、w6、w7引物。浓度为10-50μm,最优的为50μm。5、str鉴定使用双链dna染料定量上述扩增产物,取1-10ng进行str扩增试剂盒鉴定。此步骤中的str扩增试剂盒可以是以下任意一种,ab公司的identifilertm、direct、plus、sinofiler和promega公司的21system、国产试剂盒dna15plus、agcu17+1、goldeneyetm20a、阅微mr21id等。最优的为21system法医检测试剂盒。6、样本分析样本进行实验操作时最好采用一式2~5的平行对照体系,较优地采用3次,可根据实验结果进行调整和补充重复实验。str基因座位点可采用多次平行样本位点进行总结分析,避免等位基因缺失,得到的str最终位点即可进行亲权关系计算。实施例1a.具体实验步骤获得同一人单细胞样品pp-l及pp-md,同时取血液抽提基因组dna作为对照,命名为pp-b。pp-l及pp-md单细胞扩增步骤如下:1.裂解1.1将微量样本收集至1-4μl细胞收集液(1*pbs)中。1.2加入3μl细胞裂解液,总体积为7μl,不足7μl的部分使用无核酸水补齐;65℃孵育10min后,立即置于冰上。1.3加入3μl中和液,总体积为10μl。2.扩增2.1按下表2配制扩增缓冲液表22.2在pcr仪上进行如下程序:30℃孵育1-6h,65℃孵育3minutes,4℃保存。优选地30℃孵育时间为3h。2.3使用双链dna染料定量产物浓度。3.str扩增3.1取10-30ng扩增产物作为模板。3.2按照法医检测试剂盒的步骤进行操作,本次实验采用21system。3.3毛细管电泳鉴定。b.结果及分析运用id-x软件进行数据分析。单细胞检验由于其dna模板量非常低,其缺带、多带等现象经常发生,影子峰较常规扩增强,所以将分析方法的参数进行修改,将globalminusstutterratio修改为0.1,用于过滤主峰峰高10%以内的影子峰(减少一个重复单位);其余参数为默认设置。将下机数据运用id-x软件进行自动分析,人工核对内标、ladder、阴、阳性对照结果,确保每一内标峰正确、ladder每个基因座的等位基因峰均在规定范围之内,等位基因命名正确无误、阳性对照分型结果正确,阴性对照无明显目标峰。人工核查样本的分型,删除影子峰和渗透峰、人工修正双尖峰和平头峰,得到峰图如图1a、图1b、图1c;得到的分型结果如表3。pp-b为正常对照的检测结果,将pp-l、pp-md的检测结果分别与pp-b的检测结果进行比较,检测结果如表3、表4,发现存在等位基因缺失情况。等位基因缺失情况和单细胞扩增原理相关。由于正常人单细胞一般为2倍体,单细胞扩增采用随机引物pcr方法对原始模板进行扩增,一方面对原始模板的质量要求较高,任何片段的断裂或降解都有可能导致等位基因的缺失;另一方面随机引物pcr导致的偏向性始终存在,无法完全覆盖单细胞基因组全部片段,故而会存在等位基因缺失情况。为了避免等位基因缺失导致的亲权关系鉴定失败,建议之后对于微量或单细胞样本送样及检测时采用一式2~5份同源样品的平行对照体系,弥补这一方案的不足。如下表1所示,将pp-l及pp-md两个平行样本的分型结果进行总结分析,可看出所有基因座均可与pp-b匹配。说明微量或单细胞样品的str检测可与血液样本完全重合。表3pp-b与pp-l、pp-md的对比结果基因座pp-bpp-mdpp-l备注d3s135817,1917,1717,19pp-md等位基因缺失d1s165613,1613,1613,16d6s104310,1810,1810,18d13s31710,1210,1210,12pentae10,1610,1616,16pp-l等位基因缺失d16s53910,1110,1110,11d18s5114,2014,2014,20d2s133823,2523,2523,25csf1po9,109,109,10pentad12,1212,1212,12th018,98,98,9vwa14,1818,1814,18pp-md等位基因缺失d21s1130,3030,3030,30d7s82012,1212,1212,12d5s81810,1210,1210,12tpox8,98,99,9pp-l等位基因缺失d8s117915,1615,1615,16d12s39118,1818,1818,18d19s43313,1413,1413,14fga23,2423,2423,24amelx,yx,yx,y实施例2a.具体实验步骤收到单细胞样本dmd1、dmd2、fmd1、fmd2共4个。其中dmd1和2、fmd1和2分别为同一人样品,分别进行单细胞扩增,样本处理及扩增部分参考实施例一。另外单细胞扩增中设置对照组,排除扩增中引入的污染:b.结果及分析运用id-x软件进行数据分析。单细胞检验由于其dna模板量非常低,其缺带、多带等现象经常发生,影子峰较常规扩增强,所以将分析方法的参数进行修改,将globalminusstutterratio修改为0.1,用于过滤主峰峰高10%以内的影子峰(减少一个重复单位);其余参数为默认设置。将下机数据运用id-x软件进行自动分析,人工核对内标、ladder、阴、阳性对照结果,确保每一内标峰正确、ladder每个基因座的等位基因峰均在规定范围之内,等位基因命名正确无误、阳性对照分型结果正确,阴性对照无明显目标峰。人工核查样本的分型,删除影子峰和渗透峰、人工修正双尖峰和平头峰,样本dmd1、dmd2、fmd1、fmd2、单细胞扩增阴性对照样本n峰图请见图2a、图2b、图2c,图2d,图2e:其中,dmd1、dmd2、fmd1、fmd2为单个细胞样本str结果图,n为单细胞扩增阴性对照样本的单个细胞样本str结果图,得到的分型结果如表4。由以上结果看来,微量样本低至单个细胞水平-即pg级的样本经过本发明的扩增方案可有效获得str分型结果。同时阴性对照-即无dna的样本所得str结果基本为噪音,虽有部分标识为目的条带,但通过峰形可排除影响,达到排除假阳性扩增的要求。将dmd1&2、fmd1&2检测得到的位点进行合并,分别得到样本fmd、dmd相关基因座位点信息。对其进行分析,检测结果如下表:表4dmd、fmd基因座位点检测结果由上表可见,本次二联亲子鉴定共检测出了21个基因座,未出现任何矛盾基因座,本次检测父权指数达到730150.2270,可对亲子关系作出定论。以上详细描述了本发明的较佳具体实施例。应当理解,本领域的普通技术无需创造性劳动就可以根据本发明的构思作出诸多修改和变化。因此,凡本
技术领域:
中技术人员依本发明的构思在现有技术的基础上通过逻辑分析、推理或者有限的实验可以得到的技术方案,皆应在由权利要求书所确定的保护范围内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1