一种检测和分析DNA的方法与流程
本发明涉及生物技术领域,具体涉及一种检测和分析dna的方法。
背景技术:
面对快速传播的传染性疾病,迅速而准确的诊断关乎病人的生死,也与病人身边的亲属、朋友有着莫大的关系。为了降低将样品运输到专门实验室以寻找病原体或潜在突变dna所花的时间和资金成本,研究人员致力于寻找更快速准确的工具以满足这些诊断需求。以聚合酶链反应(pcr)为基础的分子诊断方法是接受最为广泛的诊断方法之一,该方法可以以相对便宜的价格获取准确、全面的诊断信息。
在试验中,可以通过探针、dna测序、毛细管电泳或dna解链曲线分析(mca)来进一步检测pcr的产物,继而进一步发掘更多的遗传信息。其中,mca的简单性和特异性使其成为了对单反应管快速检测表观遗传差异,点突变和单核苷酸多态性(snp)检测方面最有前景的分析方式。为使dna熔解,mca通常需要把温度从30℃升至90℃。dodge团队分析了来自分子信标探针的固定寡核苷酸(34-bp)的熔化过程,发现在每个温度梯度下都需要有5秒时间来使温度稳定。就同一个问题,piunno团队也对固定荧光标记的寡核苷酸双链体(20-bp)进行了熔解分析;在实验过程,试验人员在连续测量荧光的同时也对其以约0.005℃/s的速率进行加热。对于鉴别敏感的dna来说,研究人员有必要在热扫描分布过程中通过插入必要的温度稳定步骤或保持缓慢的加热速率来确保样品温度稳定和dna探针的均匀反应。尽管样品加热速率可高达175℃/s,但是这些步骤仍会减缓整个熔化过程。从上可以看出,目前用于表达dna溶解曲线的方式为加热温度与dna荧光亮度的曲线,使用该方法需要在观测过程中实时对加热温度进行监控和校准,会减缓dna解链过程,分析过程也比较繁琐。
技术实现要素:
本发明的目的在于提供一种不需要观测加热温度的检测dna的方法。
本发明的目的是通过如下技术方案实现的:
一种检测dna的方法,其包括如下步骤:
获取对照组dna的解链时间-荧光亮度曲线;
获取检测样品dna的解链时间-荧光亮度曲线;
将对照组dna的解链时间-荧光亮度曲线与检测样品dna的解链时间-荧光亮度曲线进行比较,获得检测结果。
特别地,所述对照组为空白对照组。
特别地,所述对照组为标准dna。
特别地,所述对照组为已知突变类型的dna。
特别地,在反应溶液中放入分子信标探针和dna靶,对反应溶液进行加温,同时,对其荧光信号进行记录,获取dna的解链时间-荧光亮度曲线。
特别地,在获取对照组dna和获取检测样品dna的解链时间-荧光亮度曲线过程中,两者加温过程的加温曲线保持一致。
特别地,对其荧光信号进行实时记录。
特别地,该方法用于dna突变检测或dna同源检测或目标dna有无检测。
一种检测dna的方法,其特征在于,通过获取检测样品dna的解链时间-荧光亮度曲线获得有或无目标dna的检测结果。
本发明的另一个目的在于提供一种不需要观测加热温度的分析dna的方法。
本发明的另一个目的是通过如下技术方案实现的:
一种通过获取dna的解链时间-荧光亮度曲线分析dna的方法。
特别地,在反应溶液中放入分子信标探针和dna靶,对反应溶液进行加温,同时,对其荧光信号进行记录,获取dna的解链时间-荧光亮度曲线。
本发明通过获取dna的解链时间-荧光亮度曲线来检测和分析dna,相比目前用于检测和分析dna的温度-荧光亮度曲线,不需要实时对加热温度进行精确监控和校准,加快了其分析过程,使得dna检测和分析过程变得简便和快捷。
附图说明
图1为根据本发明实施dna检测分析的原理流程图;
图2为实施例一实施dna检测和分析的流程图。
图3为实施例一所对应的空白样品和检测样品的时间-荧光亮度曲线;
图4为实施例二实施dna检测和分析的流程图。
图5为实施例二所对应的检测样品的时间-荧光亮度曲线;
图6为实施例三实施dna检测和分析的流程图。
图7为实施例三所对应的标准dna和检测样品的时间-荧光亮度曲线;
图8为实施例四实施dna检测和分析的流程图。
图9为实施例四所对应的检测样品(不同变异位点数量)的时间-荧光亮度曲线;
图10为实施例五实施dna检测和分析的流程图。
图11为实施例五所对应的检测样品(变异位点不同类型)的时间-荧光亮度曲线;
图12为实施例一、二、三、四、五所对应的加热过程的加温曲线。
具体实施方式
下面,将参照附图详细地描述本发明的具体实现原理。
如图1所示,为本发明实施检测和分析dna方法的原理。图1所示的左侧框110,是先对待测样品111(对应配对的分子信标探针的样品)按照设定的温度加热曲线112(可以是如图12所示的加热曲线)进行加热。在加热的过程中,对样品的荧光亮度被实时记录,以获取时间-荧光亮度曲线113。
观察获得的时间-荧光亮度曲线113中荧光亮度随时间的变化程度,若变化不明显则表示,待测样品中无目标基因序列,或目标基因序列数量过少,或目标基因序列基因变异位点过多115,若变化明显则表示测试样品中含有目标基因序列116。
如图1所示的右侧框120是在dna检测的基础上,继续对有目标基因序列116的待测样品进行分析。
标准样品【0】121是含有无任何变异的目标dna和对应配对的分子信标探针的样品。对标准样品【0】121加热使用的加热曲线122与对待检测样品的加温过程中温度加热曲线112一致。在加热的过程中,对样品的荧光亮度被实时记录,以获取时间-荧光亮度曲线123。对比标准样品【0】121的时间-荧光亮度曲线123和含有目标基因序列116的待测样品111的时间-荧光亮度曲线113。若两组曲线重叠,则表示待测样品111中所含有的基因序列与标准样品【0】121中含有的基因序列一致,同样表明待测样品111中所含有的基因序列中没有基因变异,为wildtype124。若两组曲线不重叠,则表示待测样品111中含有的基因序列有基因变异,为mutanttype125。
在此过程中,如果将标准样品【0】121换成空白对照样品,就可以用来确定待测样品中无目标基因序列,或目标基因序列数量过少,或目标基因序列基因变异位点过多,若变化明显则表示测试样品中含有目标基因序列。
标准样品【n】126是含有特定变异的目标dna和对应配对的分子信标探针的样品组。分别对标准样品【n】126中的样品加热,加热使用的加热曲线127与对待检测样品的加温过程中温度加热曲线112一致。在加热的过程中,对样品的荧光亮度被实时记录,以获取时间-荧光亮度曲线组128。获取的时间-荧光亮度曲线组128中抽取比对含有基因变异125的待测样品111对应的时间-荧光亮度曲线113。比对结果匹配可确认待测样品111中的目标基因的变异位点数量,或变异位点的碱基类型128。
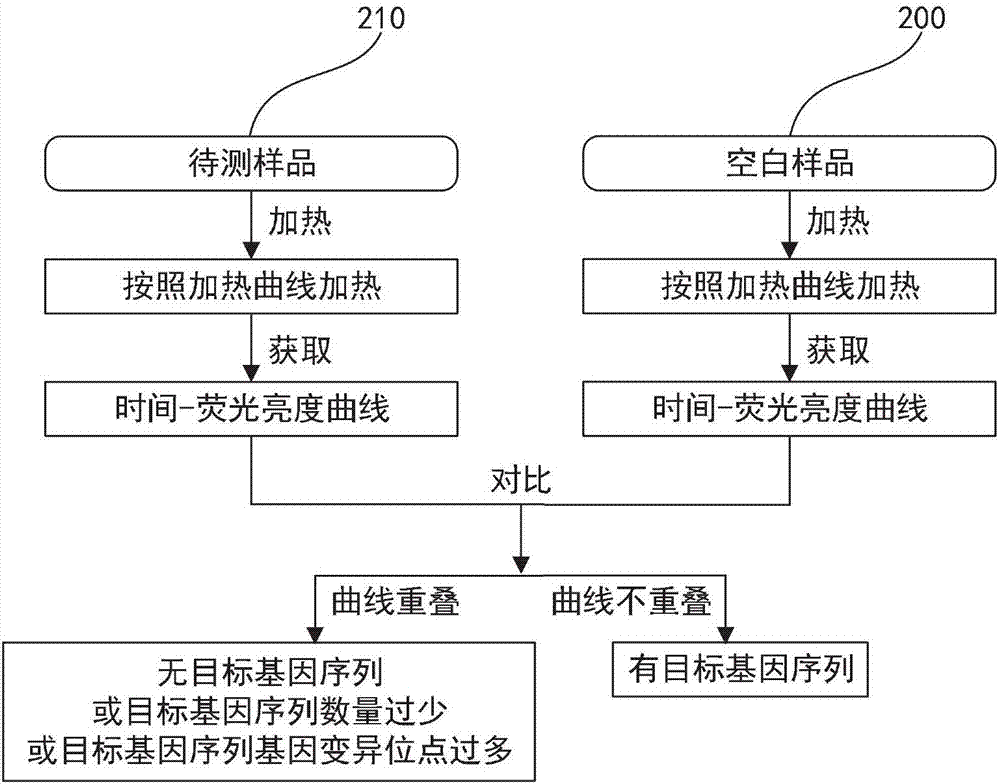
实施例一
一种检测和分析是否存在目标dna的方法,实验方法原理如图2所示,即分别对待测样品、空白样品进行加温。
实验准备:
1、取待测样品210,并在待测样品中只加入了足够数量的设计的分子信标探针5’-cy3-tctacgccaccagctca-bhq2-3’。
2、取空白样品200,并在空白样品中加入了足够数量的设计的分子信标探针5’-cy3-tctacgccaccagctca-bhq2-3’。
3、准备可对加热温度曲线进行控制的加温设备。
4、准备荧光亮度检测设备。
实验并记录:
分别对上述样品210、200进行加温,加温方式均采用图12所述的加温曲线1200(当然可以采用其他加温曲线,只要在对比测试中待测样品与空白样品保持加温曲线一致即可),并使用荧光亮度检测设备分别对样品210、200对应的荧光亮度实时记录。得到如图3所示的时间-荧光亮度曲线,其中,待测样品210对应的解链时间-荧光亮度曲线为310,空白样品200对应的解链时间-荧光亮度曲线为300。
结论
将空白样品的解链时间-荧光亮度曲线300与待检测样品dna的解链时间-荧光亮度曲线310进行比较。可以得到解链时间-荧光亮度曲线310对应检测样品210不含有目标dna或所含目标dna数量太少或所含目标dna变异位点过多。
实施例二
一种检测和分析是否存在目标dna的方法,实验方法原理如图4所示,即对待测样品进行加温。
实验准备:
1、取待测样品400,并在待测样品中只加入了足够数量的设计的分子信标探针5’-cy3-tctacgccaccagctca-bhq2-3’。
2、准备可对加热温度曲线进行控制的加温设备。
3、准备荧光亮度检测设备。
实验并记录:
分别对上述待测样品400进行加温,加温方式均采用图12所述的加温曲线1200(当然可以采用其他加温曲线),并使用荧光显微镜对待测样品400对应的荧光亮度实时记录。得到如图5所示的时间-荧光亮度曲线,待测样品400对应的解链时间-荧光亮度曲线为500。
结论
观察解链时间-荧光亮度曲线500可以得到,解链时间-荧光亮度曲线500对应检测样品400含有目标dna。
实施例三
一种检测和分析dna是否同源的方法,实验方法原理如图6所示,即分别对待测样品、标准dna(未发生任何突变)样品进行加温。
实验准备:
1、取标准dna样品600,并在标准dna样品中加入了人类kras基因片段单链dna5’-gtagttggagctggtggcgtaggcaagagt-3’及足够数量的设计的分子信标探针5’-cy3-tctacgccaccagctca-bhq2-3’;
2、取待测样品610,并在待测样品中加入了人类kras基因片段单链dna5’-gtagttggagctggtggcgtaggcaagagt-3’及足够数量的设计的分子信标探针5’-cy3-tctacgccaccagctca-bhq2-3’;
3、取待测样品620,并在待测样品中只加入了足够数量的设计的分子信标探针5’-cy3-tctacgccaccagctca-bhq2-3’。
4、准备可对加热温度曲线进行控制的加温设备。
5、准备荧光亮度检测设备。
实验并记录:
分别对上述样品610、620、600进行加温,加温方式均采用图12所述的加温曲线1200(当然可以采用其他加温曲线,只要在对比测试中待测样品与标准dna样品保持加温曲线一致即可),并使用荧光亮度检测设备分别对样品610、620、600对应的荧光亮度实时记录。得到如图7所示的时间-荧光亮度曲线,其中,样品610对应的解链时间-荧光亮度曲线为710,样品620对应的解链时间-荧光亮度曲线为720,样品600对应的解链时间-荧光亮度曲线为700。
结论
将标准dna样品的解链时间-荧光亮度曲线700与待检测样品dna的解链时间-荧光亮度曲线710、720进行比较。可以得到,解链时间-荧光亮度曲线710对应检测样品610含有目标dna,解链时间-荧光亮度曲线720对应检测样品620不含有目标dna或所含目标dna数量太少或所含目标dna变异位点过多。
实施例四
一种检测和分析dna基因突变的位点数量的方法,实验方法原理如图8所示,即分别对待测样品(确定已发生基因突变)进行加温。
实验准备:
1、取已知基因突变类型的dna样品810a,并在已知基因突变类型的dna样品810a中加入了设计的单链dna5′-atcgattagggtgtccagcgcgagcggtgggctagctcat-3′及足够数量的设计的分子信标探针5′-cy5-tagccctccgctcccgcaggccacgcta-bhq2-3′;所述设计的单链dna5′-atcgattagggtgtccagcgcgagcggtgggctagctcat-3′与计的分子信标探针5’-cy5-tagccctccgctcccgcaggccacgcta-bhq2-3′之间有5个基因突变位点。
2、取待测样品810b,并在待测样品810b中加入了设计的单链dna5′-atcgattagggtgtccagcgcgagcggtgggctagctcat-3′及足够数量的设计的分子信标探针5′-cy5-tagccctccgctcccgcaggccacgcta-bhq2-3′;
3、取已知基因突变类型的dna样品820a,并在已知基因突变类型的dna样品820a中加入了设计的单链dna5′-atcgattagggtgtccagcgcgcgcggtgggctagctcat-3′及足够数量的设计的分子信标探针5′-cy5-tagccctccgctcccgcaggccacgcta-bhq2-3′;所述设计的单链dna5′-atcgattagggtgtccagcgcgcgcggtgggctagctcat-3′与计的分子信标探针5’-cy5-tagccctccgctcccgcaggccacgcta-bhq2-3′之间有6个基因突变位点。
4、取待测样品820b,并在待测样品820b中加入了设计的单链dna5′-atcgattagggtgtccagcgcgcgcggtgggctagctcat-3′及足够数量的设计的分子信标探针5′-cy5-tagccctccgctcccgcaggccacgcta-bhq2-3′;
5、取已知基因突变类型的dna样品830a,并在已知基因突变类型的dna样品830a中加入了设计的单链dna5′-atcgattagggtgtccagcgcgcgtggtgggctagctcat-3′及足够数量的设计的分子信标探针5′-cy5-tagccctccgctcccgcaggccacgcta-bhq2-3′;所述设计的单链dna5′-atcgattagggtgtccagcgcgcgtggtgggctagctcat-3′与计的分子信标探针5’-cy5-tagccctccgctcccgcaggccacgcta-bhq2-3′之间有7个基因突变位点。
6、取待测样品830b,并在待测样品830b中加入了设计的单链dna5′-atcgattagggtgtccagcgcgcgcggtgggctagctcat-3′及足够数量的设计的分子信标探针5′-cy5-tagccctccgctcccgcaggccacgcta-bhq2-3′。
7、准备可对加热温度曲线进行控制的加温设备。
8、准备荧光亮度检测设备。
实验并记录:
分别对上述样品810a、810b、820a、820b、830a、830b、840进行加温,加温方式均采用图12所述的加温曲线1200(当然可以采用其他加温曲线,只要在对比测试中待测样品与基因突变类型的dna样品保持加温曲线一致即可),并使用荧光亮度检测设备分别对样品810a、810b、820a、820b、830a、830b、840对应的荧光亮度实时记录。得到如图9所示的时间-荧光亮度曲线,其中,样品810a对应的解链时间-荧光亮度曲线为910a,样品810b对应的解链时间-荧光亮度曲线为910b,样品820a对应的解链时间-荧光亮度曲线为920a,样品820b对应的解链时间-荧光亮度曲线为920b,样品830a对应的解链时间-荧光亮度曲线为930a,样品830b对应的解链时间-荧光亮度曲线为930b。
结论
将已知基因突变类型的dna样品的解链时间-荧光亮度曲线910a、920a、920a与待检测样品dna的解链时间-荧光亮度曲线910b、920b、930b进行比较,对应曲线高度重合,同时将对应曲线的数值放入皮尔逊相关系数公式可得对应曲线的相关性为0.999。可以得到:
解链时间-荧光亮度曲线910b对应检测样品(编号810b)中的目标dna有5个基因突变位点;
解链时间-荧光亮度曲线920b对应检测样品(编号820b)中的目标dna有6个基因突变位点;
解链时间-荧光亮度曲线930b对应检测样品(编号830b)中的目标dna有7个基因突变位点。
实施例五
一种检测和分析dna基因突变的位点数量和变异碱基位点的碱基类型的方法,实验方法原理如图10所示,即分别对待测样品(确定已发生基因突变)、已知基因突变类型的dna样品进行加温。
实验准备:
1、取已知基因突变类型的dna样品1010a,并在已知基因突变类型的dna样品1010a)中加入了设计的单链dna5′-gtagttggagctgatggcgtaggcaagagt-3′及足够数量的设计的分子信标探针5′-cy3-tctacgccaccagctca-bhq2-3′;所述设计的单链dna5′-gtagttggagctg
2、取待测样品1010b,并在待测样品1010b中加入了设计的单链dna5′-gtagttggagctgatggcgtaggcaagagt-3′及足够数量的设计的分子信标探针5′-cy3-tctacgccaccagctca-bhq2-3′;
3、取已知基因突变类型的dna样品1020a,并在已知基因突变类型的dna样品1020a中加入了设计的单链dna5′-gtagttggagctgttggcgtaggcaagagt-3′及足够数量的设计的分子信标探针5′-cy3-tctacgccaccagctca-bhq2-3′;所述设计的单链dna5′-gtagttggagctg
4、取待测样品1020b,并在待测样品1020b中加入了设计的单链dna5′-gtagttggagctgttggcgtaggcaagagt-3′及足够数量的设计的分子信标探针5′-cy3-tctacgccaccagctca-bhq2-3′;
5、取已知基因突变类型的dna样品1030a,并在已知基因突变类型的dna样品1030a中加入了设计的单链dna5′-gtagttggagctgctggcgtaggcaagagt-3′及足够数量的设计的分子信标探针5′-cy3-tctacgccaccagctca-bhq2-3′;所述设计的单链dna5′-gtagttggagctg
6、取待测样品1030b,并在待测样品1030b中加入了设计的单链dna5′-gtagttggagctgctggcgtaggcaagagt-3′及足够数量的设计的分子信标探针5′-cy3-tctacgccaccagctca-bhq2-3′。
7、准备可对加热温度曲线进行控制的加温设备。
8、准备荧光亮度检测设备。
实验并记录:
分别对上述样品1010a、1010b、1020a、1020b、1030a、1030b进行加温,加温方式均采用图12所述的加温曲线1200(当然可以采用其他加温曲线,只要在对比测试中待测样品与基因突变类型的dna样品保持加温曲线一致即可),并使用荧光亮度检测设备分别对样品1010a、1010b、1020a、1020b、1030a、1030b对应的荧光亮度实时记录。得到如图11所示的时间-荧光亮度曲线,其中,样品1010a对应的解链时间-荧光亮度曲线为1110a,样品1010b对应的解链时间-荧光亮度曲线为1110b,样品1020a对应的解链时间-荧光亮度曲线为1120a,样品1020b对应的解链时间-荧光亮度曲线为1120b,样品1030a对应的解链时间-荧光亮度曲线为1130a,样品1030b对应的解链时间-荧光亮度曲线为1130b,样品1040对应的解链时间-荧光亮度曲线为1140。
结论
将已知基因突变类型的dna样品的解链时间-荧光亮度曲线1110a、1120a、1120a与待检测样品dna的解链时间-荧光亮度曲线1110b、1120b、1130b进行比较,对应曲线高度重合,同时将对应曲线的数值放入皮尔逊相关系数公式可得对应曲线的相关性为0.999。可以得到:
解链时间-荧光亮度曲线1110b对应检测样品1010b中的目标dna有1个基因突变位点,且变异碱基由原来g变为a;
解链时间-荧光亮度曲线1120b对应检测样品1020b中的目标dna有1个基因突变位点,且变异碱基由原来g变为t;
解链时间-荧光亮度曲线1130b对应检测样品1030b中的目标dna有1个基因突变位点,且变异碱基由原来g变为c。
从上可知,该方法可以用于检测dna突变(mutation)及其数量、变异碱基位点的碱基类型或者单核苷酸多态性基因型分型(snp,singlenucleotidepolymorphismgenotyping)。
以上所述仅是本发明的优选实施例,本发明的保护范围并不仅局限于上述实施例,凡属于本发明思路下的技术方案均属于本发明的保护范围。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理前提下的若干改进和润饰,如改变分子信标探针为其他能够标识和标记dna特异性的物质,将标准dna、已知基因突变类型的dna样品及待测样品进行检测比对分析等,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!