一种重组人粒细胞集落刺激因子的制备方法与流程
本发明涉及一种重组人粒细胞集落刺激因子的制备方法,属于生物化学与分子生物学领域。
背景技术:
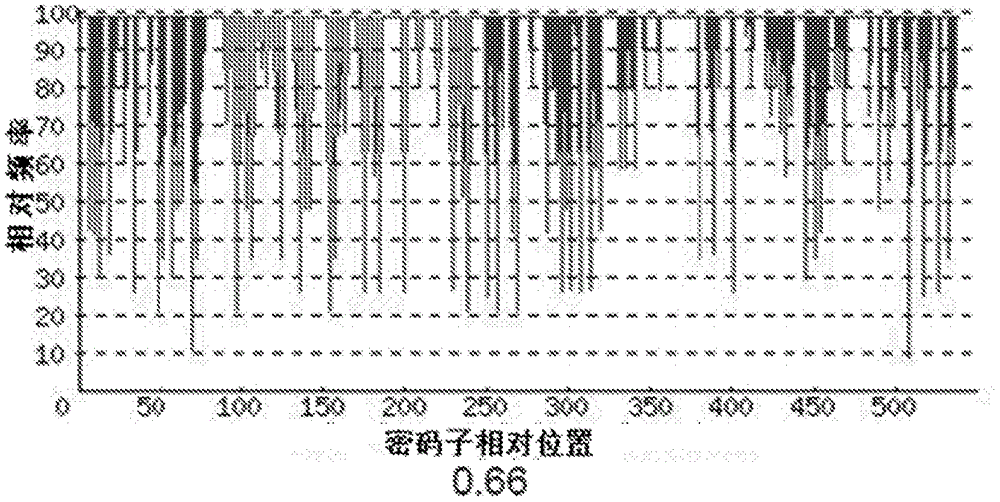
:人粒细胞集落刺激因子(hgcsf)是一种对刺激造血细胞包括成熟中性粒细胞,巨噬细胞和树状细胞的生长、分化和存活起重要作用的细胞因子。当受到微生物或者炎症细胞因子的刺激时,体内各种类型的细胞(例如:成纤维细胞,内皮细胞)都会产生人粒细胞集落刺激因子,人粒细胞集落刺激因子的活化在先天免疫应答中起重要作用。当受到特殊的抗原刺激时,t细胞也会分泌人粒细胞集落刺激因子。由于对造血细胞的促进作用,人粒细胞集落刺激因子常被用作对免疫低下个体治疗的生物药。hgcsf的活化失控与诸如关节炎和多发性硬化等自发免疫症状相关。自身抗体对hgcsf生物学活性的中和引起另一中自身免疫疾病---特发型肺泡蛋白沉积症,并且hgcsf可以治疗这种症状。目前,人粒细胞集落刺激因子主要应用于癌症放、化疗等原因引起的白细胞减少症,是肿瘤放、化疗过程中重要的辅助药物。1986年人粒细胞集落刺激因子cdna克隆成功,人粒细胞集落刺激因子基因全长2.5kb,包括5个外显子和4个内含子,人粒细胞集落刺激因子基因位于17号染色体。人类有两种不同的hgcsfcdna,分别编码含207和204个氨基酸的前体蛋白,均有30个氨基酸的先导序列,成熟蛋白分子分别为177和174个氨基酸,前者除了在成熟分子n端35位处插入了3个氨基酸外,其余的序列与174氨基酸分子相同。174氨基酸分子的生物学活性比177氨基酸分子的生物学活性高20倍。通常而言,人粒细胞集落刺激因子使用哺乳动物细胞进行重组表达。但是,为了分离和纯化hgcsf,培养细胞并从培养上清液中分离gcsf蛋白,该方法存在gcsf产量低的缺点,因此不适于大量生产。已知糖基化hgcsf的糖链对于hgcsf的活性而言不是必需的,并且使用哺乳动物细胞产生糖基化hgcsf需要昂贵的材料和设备,因此,这样的方法从经济角度上来说不可行。1991年,第一个通过大肠杆菌表达系统生产的重组人粒细胞集落刺激因子药物filgrastim(neupogen,r-methugcsf,amgeninc)在美国上市,此后国内外有多家公司的大肠杆菌表达系统生产的rhgcsf药物上市。目前报道的关于提高重组人粒细胞集落刺激因子产量的主要方法有以下几种(1)通过改变表达体系来提高表达量;(2)改变发酵工艺,如高密度发酵、优化培养基、优化培养条件等手段实现工程菌的高表达;(3)优化复性条件,更改纯化工艺。但是这些方法只是生产工艺方面的优化,并没有在分子水平上提高人粒细胞集落刺激因子的重组表达产量。如上文所述,糖基化hgcsf的糖链对于hgcsf的活性而言不是必需的,人粒细胞集落刺激因子可使用更加经济和方便的大肠杆菌表达系统进行生产,中国专利cn1156575c、cn1108303a、cn1088107c、cn101591660a等都是直接采用人源cdna序列在大肠杆菌表达系统中重组表达人粒细胞集落刺激因子,而大肠杆菌表达系统中真核蛋白的表达会出现稀有密码子问题。尽管可以使用从bl21衍生而来的rosetta宿主菌增强带有大肠杆菌稀有密码子的真核蛋白的表达。但是rosetta宿主菌的筛选标记为氯霉素抗性,在药物生产中该抗生素的使用具有较大的限制。如何提高重组人粒细胞集落刺激因子的产量,最大限度的降低成本,是个巨大的挑战。技术实现要素:本发明提供一种重组人粒细胞集落刺激因子的制备方法,该方法采用不同于重组人粒细胞集落刺激因子人源序列seqidno.1的cdna在原核表达系统中表达,通过本发明提供的方法可使目的蛋白即重组人粒细胞刺激因子的表达量显著提高,解决了直接采用人源cdna序列在原核生物表达体系中出现的稀有密码子问题。本发明提供了一种重组人粒细胞集落刺激因子的制备方法,重组人粒细胞集落刺激因子的蛋白质序列见seqidno.3,该蛋白序列对应的重组人粒细胞集落刺激因子的人源基因序列见seqidno.1,该方法的特征在于包含合成cdna序列的步骤,所述cdna由人源cdna序列突变而成,还包括重组表达载体的构建步骤以及重组表达载体在宿主细胞中诱导表达的步骤。本发明中合成cdna序列的步骤,采用全基因合成法,根据大肠杆菌密码子偏好性,同时优化序列gc含量,mrna二级结构,消除剪接位点,polya位点,chi位点和rbs位点,cpg岛,rna不稳定模体,重复序列以及可能干扰克隆的限制性酶切位点,优化重组人粒细胞集落刺激因子基因序列。为了便于载体构建,在两端分别引入克隆重组所需的的酶切位点。在一个优选的实施方案中,本发明提供的方法中合成的cdna序列见seqidno.2。本发明提供的重组人粒细胞集落刺激因子的cdna序列seqidno.2的合成是将重组人粒细胞集落刺激因子的人源基因序列中的34个碱基替换得到,序列中所有的大肠杆菌低频密码子均被高频密码子所替换。合成的cdna序列seqidno.2在大肠杆菌中的密码子适应性增加(见图1a和图1b),密码子使用频率得到提升(见图2a和图2b),此外gc含量也有所改善(见图3a和图3b)。本发明提供的重组人粒细胞集落刺激因子的制备方法还包括重组表达载体的构建的步骤。本发明提供的方法中涉及的重组表达载体选自温度表达载体(例如pbv220)和底物诱导载体(例如pet9a),优选底物诱导表达载体,最优选pet系列。本发明提供的重组人粒细胞集落刺激因子的制备方法还包括重组表达载体在宿主细胞中诱导表达的步骤。本发明提供的方法中涉及的宿主细胞为大肠杆菌,优选大肠杆菌escherichiacolidh5a、bl21(de3)、rosetta(de3)、origami(de3)、jm109、hsm174(de3)。具体来讲,本发明提供的方法包含以下步骤:(1)采用全基因合成法合成重组人粒细胞集落刺激因子基因;(2)合成的cdna序列与表达载体连接,构建重组表达载体。(3)构建的表达载体在大肠杆菌中进行高效表达。通过本发明提供的cdna序列的重组表达产量可以达到人源cdna的2倍以上。本发明还提供了一种根据上述全基因合成法合成的可编码重组人粒细胞集落刺激因子的cdna序列,见seqidno.2。本发明还提供了一种重组人粒细胞集落刺激因子基因的重组表达载体,该表达载体含有seqidno.2。本发明提供的重组表达载体选自温度表达载体(例如pbv220)和底物诱导载体(例如pet9a),优选底物诱导表达载体,最优选pet系列。本发明还提供了一种可表达重组人粒细胞集落刺激因子的宿主细胞,该宿主细胞含有上述的重组表达载体。本发明提供的宿主细胞为大肠杆菌,优选大肠杆菌escherichiacolidh5a、bl21(de3)、rosetta(de3)、origami(de3)、jm109、hsm174(de3)。本发明还提供了一种制备重组人粒细胞集落刺激因子的水溶性聚合物修饰的偶联物的方法,该方法采用前述步骤得到如蛋白质序列seqidno.3所示的重组人粒细胞集落刺激因子后,进一步包含重组人粒细胞集落刺激因子与水溶性聚合物偶联的步骤,具体操作方法可参考专利cn101172161b。本发明还提供了一种重组人粒细胞集落刺激因子的水溶性聚合物修饰的偶联物,所述的水溶性聚合物选自聚乙二醇、聚丙二醇、聚乳酸等,优选聚乙二醇,聚乙二醇的分子量选自2kd-100kd,优选5kd-100kd。本发明提供的重组人粒细胞集落刺激因子的水溶性聚合物修饰物的偶联物结构如式(i)所示:m选自50-2500的整数,优选自400-500的整数,g为序列seqidno.3所示的蛋白序列。附图说明图1a和图1b:序列seqidno.1(图1a)与序列seqidno.2(图1b)密码子适应指数(cai)对比。图2a和图2b:序列seqidno.1(图2a)与序列seqidno.2(图2b)密码子使用频率(fop)对比。图3a和图3b:序列seqidno.1(图3a)与序列seqidno.2(图3b)gc含量对比。图4:重组表达菌株的摇瓶诱导表达,其中泳道1:pet9a-gcsf-1/bl21(de3),泳道2:pet9a-gcsf-2/bl21(de3),泳道3:pet9a-gcsf-2/bl21(de3),泳道4:pbv220-gcsf-3/dh5a,泳道5:pbv220-gcsf-4/dh5a,泳道6:pbv220-gcsf-4/dh5a,泳道7:空载体菌样品,泳道8:空菌样品,泳道m:蛋白标记(proteinmarker)。图5:重组表达菌株5l发酵罐诱导表达产量测定,其中1:发酵样品稀释8倍,2:发酵样品稀释16倍。具体实施方式以下结合实施例用于进一步描述本发明,但这些实施例并非限制本发明的范围。实施例1:重组人粒细胞集落刺激因子底物诱导表达菌株的构建1、基因合成重组人粒细胞集落刺激因子基因序列由南京金斯瑞生物公司合成。重组人粒细胞集落刺激因子人源序列seqidno.1:合成的重组人粒细胞集落刺激因子基因序列gcsf-1如下所示,在seqidno.15’端加了一个ndei酶切位点,3’端添加了一个bamhi酶切位点。重组人粒细胞集落刺激因子cdna序列seqidno.2:合成的重组人粒细胞集落刺激因子基因序列gcsf-2如下所示,在seqidno.25’端加了一个ndei酶切位点,3’端添加了一个bamhi酶切位点。对应的蛋白序列如seqidno.3所示2、重组表达菌株pet9a-gcsf-1/bl21(de3)、pet9a-gcsf-2/bl21(de3)的构建采用ndei和bamhi内切酶(fermentas)分别对重组克隆质粒(puc57-gcsf-1、puc57-gcsf-2)和表达载体pet9a进行双酶切消化。采用快速胶回收试剂盒(promega)对目的基因片段(gcsf-1、gcsf-2)和表达载体pet9a酶切产物进行胶回收,用t4连接酶(newenglandbiolabs)对目的基因片段(gcsf-1、gcsf-2)和表达载体pet9a回收片段进行dna连接。最后将连接产物转化大肠杆菌bl21(de3)感受态细胞,涂布含终浓度50μg/mlkana的lb平板筛选重组子。挑取阳性克隆,抽提质粒并由南京金斯瑞公司测序验证,得到pet9a-gcsf-1/bl21(de3)、pet9a-gcsf-2/bl21(de3)表达菌株。3、重组表达菌株pet9a-gcsf-1/bl21(de3)、pet9a-gcsf-2/bl21(de3)的摇瓶诱导表达将测序验证的阳性克隆接种至含2mllb(50μg/mlkana)培养基的12ml菌种培养管中,37℃恒温摇床225rpm振荡培养至od600=4左右(16-18小时)。转接入50mllb(50μg/mlkana)培养基中,37℃恒温摇床220rpm振荡培养约1.5h至od600=0.8左右。在培养基中加入约1mmiptg,37℃恒温摇床220rpm振荡培养4-5小时,表达结束后,设置离心力为5000g离心10分钟取沉淀,sds-page电泳分析见图4。4、重组表达菌株pet9a-gcsf-1/bl21(de3)、pet9a-gcsf-2/bl21(de3)5l发酵罐诱导表达5l规模发酵分如下4步进行:1.取2ml甘油菌接种至装有200mllb培养基(酵母粉5g/l,大豆蛋白胨10g/l,nacl5g/l)的1l摇瓶中,37℃恒温摇床220rpm振荡培养约9h,培养至菌种od600=2.5左右。2.将培养好的种子液接种至5l发酵罐中进行发酵,使用3llb培养基(其组分为:胰蛋白胨10g/l,酵母粉5g/l,nacl5g/l,泡敌0.03%,na2hpo4·12h2o11g/l,kh2po42.7g/l,121℃灭菌30min。葡萄糖5g/l,mgso40.3g/l,115℃灭菌30min),培养温度设定为37℃,通气量保持6l/min,用氨水控制ph在7.0左右,37℃培养,通过搅拌和补料控制溶氧在30%~110%。3.od600达到25左右时,加入约1mmiptg,诱导时间4h。4.产量测定表1菌株诱导后2h诱导后4h诱导后6h优化前pet9a-gcsf-1/bl21(de3)1.23(g/l)1.51(g/l)1.74(g/l)优化后pet9a-gcsf-2/bl21(de3)2.36(g/l)3.04(g/l)3.49(g/l)由表1中结果可知:采用seqidno.2基因序列构建的工程菌的目的蛋白的表达量是采用人源的重组人粒细胞集落刺激因子基因序列seqidno.1构建的工程菌目的蛋白的表达量的2倍以上。实施例2:重组人粒细胞集落刺激因子温度诱导表达菌株的构建1.基因合成重组人粒细胞集落刺激因子基因序列由南京金斯瑞生物公司合成。合成的重组人粒细胞集落刺激因子基因序列gcsf-3如下所示,在seqidno.15’端加了一个ecori酶切位点,3’端添加了一个bamhi酶切位点。合成的重组人粒细胞集落刺激因子基因序列gcsf-4如下所示,在seqidno.25’端加了一个ecori酶切位点,3’端添加了一个bamhi酶切位点。对应的蛋白序列如seqidno.3所示2.重组表达菌株pbv220-gcsf-3/dh5a、pbv220-gcsf-4/dh5a的构建采用ecori和bamhi内切酶(fermentas)分别对重组克隆质粒(puc57-gcsf-3、puc57-gcsf-4)和表达载体pbv220进行双酶切消化。采用快速胶回收试剂盒(promega)对目的基因片段(gcsf-3、gcsf-4)和表达载体pbv220酶切产物进行胶回收,用t4连接酶(newenglandbiolabs)对目的基因片段(gcsf-3、gcsf-4)和表达载体pbv220回收片段进行dna连接。最后将连接产物转化大肠杆菌dh5a感受态细胞,涂布含终浓度100μg/mlamp的lb平板筛选重组子。挑取阳性克隆,抽提质粒并由南京金斯瑞公司测序验证,得到pbv220-gcsf-3/dh5a、pbv220-gcsf-4/dh5a表达菌株。3.重组表达菌株pbv220-gcsf-3/dh5a、pbv220-gcsf-4/dh5a的摇瓶诱导表达将测序验证的阳性克隆接种至含2mllb(100μg/mlamp)培养基的12ml菌种培养管中,30℃恒温摇床225rpm振荡培养至od600=4左右(16-18小时)。转接入50mllb(100μg/mlamp)培养基中,30℃恒温摇床220rpm振荡培养约1.5h至od600=0.8左右。升温至42℃恒温摇床220rpm振荡培养4-5小时,表达结束后,设置离心力为5000g离心10分钟取沉淀,sds-page电泳分析。4.重组表达菌株pbv220-gcsf-3/dh5a、pbv220-gcsf-3/dh5a5l发酵罐诱导表达5l规模发酵分如下4步进行:1.取2ml甘油菌接种至装有200mllb培养基(酵母粉5g/l,大豆蛋白胨10g/l,nacl5g/l)的1l摇瓶中,30℃恒温摇床220rpm振荡培养约9h,培养至菌种od600=2.5左右。2.将培养好的种子液接种至5l发酵罐中进行发酵,使用3llb培养基(其组分为:胰蛋白胨10g/l,酵母粉5g/l,nacl5g/l,泡敌0.03%,na2hpo4·12h2o11g/l,kh2po42.7g/l,121℃灭菌30min。葡萄糖5g/l,mgso40.3g/l,115℃灭菌30min),培养温度设定为30℃,通气量保持6l/min,用氨水控制ph在7.0左右,30℃培养,通过搅拌和补料控制溶氧在30%~110%。3.od600达到25左右时,升温至42℃,诱导时间4h。4.产量测定表2菌株诱导后2h诱导后4h诱导后6h优化前pbv220-gcsf-3/dh5a0.53(g/l)0.81(g/l)1.24(g/l)优化后pbv220-gcsf-4/dh5a1.56(g/l)2.18(g/l)2.52(g/l)由表2中结果可知:采用seqidno.2基因序列构建的工程菌的目的蛋白的表达量是采用人源的重组人粒细胞集落刺激因子基因序列seqidno.1构建的工程菌目的蛋白的表达量的2倍以上。实施例3:重组表达菌株5l发酵罐诱导表达产量的测定1、取对照品30μl,用纯化水稀释配制0.20g/l对照品溶液,进一步用纯化水稀释成浓度梯度。取1ml混合均匀的发酵液,12000rpm离心5分钟,弃上清,沉淀中加入1ml纯化水混匀。取52μl稀释样品,加入20μlldssamplebuffer(4×)和8μlreducingagent(10×),混匀,70℃水浴10分钟,12000rpm离心1分钟备用。2、选择4-12%bis-trisgel与mopssdsrunningbuffer(20×)与200v恒压电泳约50分钟至溴酚蓝迁移至胶底,停止电泳。电泳完毕,取出胶片,置约100ml纯化水中,摇床振荡5min,重复三次,弃去纯化水。加入约100ml染色液,摇床振荡染色1h直到出现清晰的蛋白条带;弃去染色液,加入约100ml纯化水,摇床振荡脱色过夜至凝胶背景透明。3、将电泳后的凝胶置凝胶成像仪中,拍照分析见图5。实施例4:重组人粒细胞集落刺激因子cdna序列筛选1、基因合成重组人粒细胞集落刺激因子基因序列由南京金斯瑞生物公司合成。重组人粒细胞集落刺激因子cdna序列seqidn0.4:合成的重组人粒细胞集落刺激因子基因序列gcsf-5如下所示,在seqidno.45’端加了一个ndei酶切位点,3’端添加了一个bamhi酶切位点。重组人粒细胞集落刺激因子cdna序列seqidno.5:合成的重组人粒细胞集落刺激因子基因序列gcsf-6如下所示,在seqidno.55’端加了一个ndei酶切位点,3’端添加了一个bamhi酶切位点。对应的蛋白序列如seqidno.3所示2、重组表达菌株pet9a-gcsf-5/bl21(de3)、pet9a-gcsf-6/bl21(de3)的构建采用ndei和bamhi内切酶(fermentas)分别对重组克隆质粒(puc57-gcsf-5、puc57-gcsf-6)和表达载体pet9a进行双酶切消化。采用快速胶回收试剂盒(promega)对目的基因片段(gcsf-5、gcsf-6)和表达载体pet9a酶切产物进行胶回收,用t4连接酶(newenglandbiolabs)对目的基因片段(gcsf-5、gcsf-6)和表达载体pet9a回收片段进行dna连接。最后将连接产物转化大肠杆菌bl21(de3)感受态细胞,涂布含终浓度50μg/mlkana的lb平板筛选重组子。挑取阳性克隆,抽提质粒并由南京金斯瑞公司测序验证,得到pet9a-gcsf-5/bl21(de3)、pet9a-gcsf-6/bl21(de3)表达菌株。3、重组表达菌株pet9a-gcsf-1/bl21(de3)、pet9a-gcsf-2/bl21(de3)、pet9a-gcsf-5/bl21(de3)、pet9a-gcsf-6/bl21(de3)的摇瓶诱导表达将测序验证的阳性克隆接种至含2mllb(50μg/mlkana)培养基的12ml菌种培养管中,37℃恒温摇床225rpm振荡培养至od600=4左右(16-18小时)。转接入50mllb(50μg/mlkana)培养基中,37℃恒温摇床220rpm振荡培养约1.5h至od600=0.8左右。在培养基中加入约1mmiptg,37℃恒温摇床220rpm振荡培养4-5小时,表达结束后,设置离心力为5000g离心10分钟取沉淀,sds-page电泳分析,按实施例3所示方法进行定量,结果见下表:表3:当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1