具有改善的集结粒流动性的乙烯/C3-C6α-烯烃互聚物的制作方法
本申请案要求2016年8月30日申请的国际申请案pct/us16/049458的优先权。
背景技术:
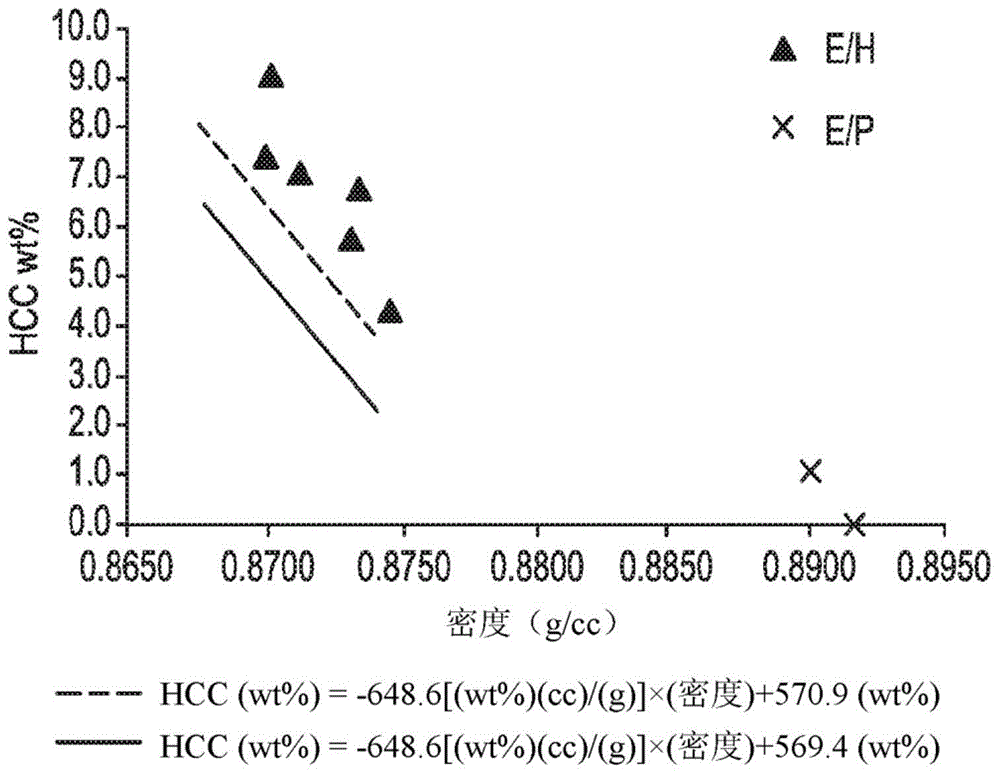
:低分子量乙烯/α-烯烃互聚物通常在由这类互聚物形成的集结粒的流动期间呈现粘性。需要具有降低的粘连(粘着)特征的乙烯/c3-c6α-烯烃互聚物,并且其可以用于粘着剂配制物中。这些需求已经通过本发明满足。技术实现要素:提供一种组合物,其包含乙烯/c3-c6α-烯烃互聚物,并且其中所述互聚物包含以下特性:a)符合以下方程式的hcc值:hcc(重量%)≥-648.6[(重量%)(cc)/(g)]×(密度)+569.4(重量%);b)老化流动性≥130克/秒。附图说明图1描绘若干种本发明的乙烯/c3-c6α-烯烃互聚物的“hcc与密度”曲线。图2描绘若干种本发明的乙烯/c3-c6α-烯烃互聚物的“hcc与密度”曲线。图3描绘用于集结粒流动性研究的测定漏斗。图4是用于测定比较性共聚物的hcc值的实例色谱图。图5描绘用于tgic温度校准的洗脱温度的外推法。图6展示聚合物eh-2和eh-7的ht-tgic层析图(第2号hcc方法)。具体实施方式如上文所讨论,提供一种组合物,其包含乙烯/c3-c6α-烯烃互聚物,并且其中所述互聚物包含以下特性:a)符合以下方程式的hcc值:hcc(重量%)≥-648.6[(重量%)(cc)/(g)]×(密度)+569.4(重量%);b)老化流动性≥130克/秒。一种所提供的组合物可以包含如本文中所述的两个或更多个实施例的组合。在一个实施例中,组合物呈集结粒形式。在一个实施例中,使用hcc方法2测定hcc值。在一个实施例中,乙烯/c3-c6α-烯烃互聚物具有符合以下方程式的hcc值:hcc(重量%)≥-648.6[(重量%)(cc)/(g)]×(密度)+570.9(重量%)。在另一实施例中,α-烯烃是c3或c6α-烯烃。在一个实施例中,使用hcc方法2测定hcc值。在一个实施例中,乙烯/c3-c6α-烯烃互聚物进一步包含符合以下方程式的hcc值:hcc(重量%)<-463.4[(重量%)(cc)/(g)]×(密度)+413.8(重量%)。在另一实施例中,α-烯烃是c3或c6α-烯烃。在一个实施例中,使用hcc方法2测定hcc值。在一个实施例中,乙烯/c3-c6α-烯烃互聚物的hcc值≥0重量%,或≥1.0重量%,或≥4.0重量%,或≥4.2重量%,或≥4.5重量%,或≥5.0重量%,或≥5.5重量%,或≥6.0重量%。在另一实施例中,α-烯烃是c3或c6α-烯烃。在一个实施例中,使用hcc方法2测定hcc值。在一个实施例中,乙烯/c3-c6α-烯烃互聚物的hcc值≤12.0重量%,或≤11.5重量%,或≤11.0重量%,或≤10.5重量%,或≤10.0重量%,或≤9.5重量%,或≤9.0重量%。在另一实施例中,α-烯烃是c3或c6α-烯烃。在一个实施例中,使用hcc方法2测定hcc值。在一个实施例中,乙烯/c3-c6α-烯烃互聚物的hcc值≥4.0重量%,或≥4.2重量%,或≥4.5重量%,或≥5.0重量%,或≥5.5重量%,或≥6.0重量%。在另一实施例中,互聚物是乙烯/c6α-烯烃共聚物。在一个实施例中,使用hcc方法2测定hcc值。在一个实施例中,乙烯/α-烯烃互聚物的hcc值≤11.0重量%,或≤10.5重量%,或≤10.0重量%,或≤9.5重量%,或≤9.0重量%。在另一实施例中,互聚物是乙烯/c6α-烯烃共聚物。在一个实施例中,使用hcc方法2测定hcc值。在一个实施例中,乙烯/c3-c6α-烯烃互聚物的hcc值≥0重量%,或≥0.5重量%,或≥1.0重量%。在另一实施例中,互聚物是乙烯/c3α-烯烃互聚物,或共聚物。在一个实施例中,使用hcc方法2测定hcc值。在一个实施例中,乙烯/α-烯烃互聚物的hcc值≤5.0重量%,或≤4.5重量%,或≤4.0重量%,或≤3.5重量%,或≤3.0重量%。在另一实施例中,互聚物是乙烯/c3α-烯烃互聚物,或共聚物。在一个实施例中,使用hcc方法2测定hcc值。hcc(或“高共聚单体含量”)值是在聚合期间形成的聚合物的组成特征,并且不是残余杂质。hcc值指示聚合物样品中低密度、低分子量、低聚组分的量。hcc值不表示全部聚合物样品中的平均共聚单体含量。参见以下用于测定hcc值的测试方法。分析全部聚合物样品并且非溶剂萃取部分的hcc值。使用色谱测试方法测量聚合物样品中的共聚单体分布,并且不用于测量聚合物样品中的杂质。“共聚单体分布”是聚合物并且非杂质的微观结构参数。在一个实施例中,使用hcc方法2测定hcc值。在一个实施例中,乙烯/c3-c6α-烯烃互聚物的老化流动性≥135.0克/秒,或≥140.0克/秒,或≥145.0克/秒,或≥150.0克/秒,或≥155.0克/秒。在另一实施例中,α-烯烃是c3或c6α-烯烃。通过本文中所描述的方法测定老化流动性。在一个实施例中,组合物呈集结粒形式。在一个实施例中,乙烯/c3-c6α-烯烃互聚物的老化流动性≤400.0克/秒,或≤350.0克/秒,或≤300.0克/秒。在另一实施例中,α-烯烃是c3或c6α-烯烃。通过本文中所描述的方法测定老化流动性。在一个实施例中,组合物呈集结粒形式。在一个实施例中,乙烯/c3-c6α-烯烃互聚物的老化流动性≥135.0克/秒,或≥140.0克/秒,或≥145.0克/秒。在另一实施例中,互聚物是乙烯/c6α-烯烃共聚物。在一个实施例中,乙烯/c3-c6α-烯烃互聚物的老化流动性≤500.0克/秒,或≤400.0克/秒,或≤300.0克/秒。在另一实施例中,互聚物是乙烯/c6α-烯烃共聚物。通过本文中所描述的方法测定老化流动性。在一个实施例中,组合物呈集结粒形式。在一个实施例中,乙烯/c3-c6α-烯烃互聚物的老化流动性≥140.0克/秒,或≥150.0克/秒,或≥160.0克/秒。在另一实施例中,互聚物是乙烯/c3α-烯烃互聚物,或共聚物。在一个实施例中,乙烯/c3-c6α-烯烃互聚物的老化流动性≤450.0克/秒,或≤400.0克/秒,或≤350.0克/秒。在另一实施例中,互聚物是乙烯/c3α-烯烃互聚物,或共聚物。通过本文中所描述的方法测定老化流动性。在一个实施例中,组合物呈集结粒形式。在一个实施例中,乙烯/c3-c6α-烯烃互聚物在350℉(177℃)下的熔融粘度≤50,000cp,或≤45,000cp,或≤40,000cp,或≤35,000cp,或≤30,000cp,或≤25,000cp,或≤20,000cp。在另一实施例中,α-烯烃是c3或c6α-烯烃。在另一实施例中,α-烯烃是c3或c6α-烯烃。在另一实施例中,互聚物是乙烯/c6α-烯烃共聚物。在另一实施例中,互聚物是乙烯/c3α-烯烃互聚物,或共聚物。在一个实施例中,乙烯/c3-c6α-烯烃互聚物在350℉(177℃)下的熔融粘度≥2,000cp,或≥2,500cp,或≥3,000cp,或≥3,500cp,或≥4,000cp,或≥4,500cp,或≥5,000cp,或≥5,500cp,或≥6,000cp。在另一实施例中,α-烯烃是c3或c6α-烯烃。在另一实施例中,α-烯烃是c3或c6α-烯烃。在另一实施例中,互聚物是乙烯/c6α-烯烃共聚物。在另一实施例中,互聚物是乙烯/c3α-烯烃互聚物,或共聚物。在一个实施例中,乙烯/c3-c6α-烯烃互聚物在350℉(177℃)下的熔融粘度是2,000cp到50,000cp,或2,500cp到45,000,或3,000cp到40,000,或3,500cp到35,000,或4,000cp到30,000,或4,500cp到25,000,或5,000cp到20,000。在另一实施例中,α-烯烃是c3或c6α-烯烃。在另一实施例中,互聚物是乙烯/c6α-烯烃共聚物。在另一实施例中,互聚物是乙烯/c3α-烯烃互聚物,或共聚物。在一个实施例中,乙烯/c3-c6α-烯烃互聚物的密度≥0.855g/cc,或≥0.860g/cc,或≥0.865g/cc,或≥0.870g/cc(1cc=1cm3)。在另一实施例中,α-烯烃是c3或c6α-烯烃。在一个实施例中,乙烯/c3-c6α-烯烃互聚物的密度≤0.910g/cc,或≤0.905,或≤0.900g/cc,或≤0.895g/cc。在另一实施例中,α-烯烃是c3或c6α-烯烃。在一个实施例中,乙烯/c3-c6α-烯烃互聚物的密度是0.855到0.900g/cc,或0.860到0.895g/cc,或0.865到0.890g/cc,或0.870到0.885g/cc。在另一实施例中,互聚物是乙烯/c6α-烯烃共聚物。在一个实施例中,乙烯/c3-c6α-烯烃互聚物的密度是0.875到0.910g/cc,或0.880到0.905g/cc,或0.885到0.900g/cc,或0.890到0.895g/cc。在另一实施例中,互聚物是乙烯/c3α-烯烃互聚物,或共聚物。在一个实施例中,乙烯/c3-c6α-烯烃互聚物的最高峰值熔融温度≤85.0℃,或≤82.0℃,或≤80.0℃。在另一实施例中,α-烯烃是c3或c6α-烯烃。在另一实施例中,互聚物是乙烯/c6α-烯烃共聚物。在另一实施例中,互聚物是乙烯/c3α-烯烃互聚物,或共聚物。在一个实施例中,乙烯/c3-c6α-烯烃互聚物的最高峰值熔融温度≥60.0℃,或≥62.0℃,或≥64.0℃。在另一实施例中,α-烯烃是c3或c6α-烯烃。在另一实施例中,互聚物是乙烯/c6α-烯烃共聚物。在另一实施例中,互聚物是乙烯/c3α-烯烃互聚物,或共聚物。在一个实施例中,乙烯/c3-c6α-烯烃互聚物的最高峰值熔融温度≤80.0℃,或≤78.0℃,或≤76.0℃。在另一实施例中,互聚物是乙烯/c6α-烯烃共聚物。在一个实施例中,乙烯/α-烯烃互聚物的最高峰值熔融温度≥60.0℃,或≥62.0℃,或≥64.0℃。在另一实施例中,互聚物是乙烯/c6α-烯烃共聚物。在一个实施例中,乙烯/c3-c6α-烯烃互聚物的熔融温度≤84.0℃,或≤82.0℃,或≤80.0℃。在另一实施例中,互聚物是乙烯/c3α-烯烃互聚物。在一个实施例中,乙烯/α-烯烃互聚物的熔融温度≥72.0℃,或≥74.0℃,或≥76.0℃。在另一实施例中,互聚物是乙烯/c3α-烯烃互聚物,或共聚物。在一个实施例中,乙烯/c3-c6α-烯烃互聚物的结晶温度(tc)≤72.0℃,或≤70.0℃,或≤68.0℃。在另一实施例中,α-烯烃是c3或c6α-烯烃。在另一实施例中,互聚物是乙烯/c6α-烯烃共聚物。在另一实施例中,互聚物是乙烯/c3α-烯烃互聚物,或共聚物。在一个实施例中,乙烯/c3-c6α-烯烃互聚物的结晶温度(tc)≥48.0℃,或≥50.0℃,或≥52.0℃。在另一实施例中,α-烯烃是c3或c6α-烯烃。在另一实施例中,互聚物是乙烯/c6α-烯烃共聚物。在另一实施例中,互聚物是乙烯/c3α-烯烃互聚物,或共聚物。在一个实施例中,乙烯/c3-c6α-烯烃互聚物的结晶温度(tc)≤70.0℃,或≤68.0℃,或≤66.0℃。在另一实施例中,互聚物是乙烯/c6α-烯烃共聚物。在一个实施例中,乙烯/α-烯烃互聚物的结晶温度(tc)≥48.0℃,或≥50.0℃,或≥52.0℃。在另一实施例中,互聚物是乙烯/c6α-烯烃共聚物。在一个实施例中,乙烯/c3-c6α-烯烃互聚物的结晶温度(tc)≤72.0℃,或≤70.0℃,或≤68.0℃。在另一实施例中,互聚物是乙烯/c3α-烯烃互聚物,或共聚物。在一个实施例中,乙烯/α-烯烃互聚物的结晶温度(tc)≥58.0℃,或≥60.0℃,或≥62.0℃。在另一实施例中,互聚物是乙烯/c3α-烯烃互聚物,或共聚物。在一个实施例中,乙烯/c3-c6α-烯烃互聚物的tm和tc满足以下关系:(tm-tc)是5℃到25℃,进一步是8℃到20℃,进一步是10℃到15℃。在另一实施例中,α-烯烃是c3或c6α-烯烃。在另一实施例中,互聚物是乙烯/c6α-烯烃共聚物。在另一实施例中,互聚物是乙烯/c3α-烯烃互聚物,或共聚物。在一个实施例中,乙烯/c3-c6α-烯烃互聚物的结晶度百分比≤50%,或≤45%,或≤40%,或≤35%,或≤30%,如通过dsc测定。在另一实施例中,α-烯烃是c3或c6α-烯烃。在另一实施例中,互聚物是乙烯/c6α-烯烃共聚物。在另一实施例中,互聚物是乙烯/c3α-烯烃互聚物,或共聚物。在一个实施例中,乙烯/c3-c6α-烯烃互聚物的结晶度百分比≥8%,或≥10%,或≥12%,或≥15%,或≥17%,如通过dsc测定。在另一实施例中,α-烯烃是c3或c6α-烯烃。在另一实施例中,互聚物是乙烯/c6α-烯烃共聚物。在另一实施例中,互聚物是乙烯/c3α-烯烃互聚物,或共聚物。在一个实施例中,乙烯/c3-c6α-烯烃互聚物的结晶度百分比≤35%,或≤30%,或≤25%。在另一实施例中,互聚物是乙烯/c6α-烯烃共聚物。在一个实施例中,乙烯/α-烯烃互聚物的结晶度百分比≥12%,或≥14%,或≥16%。在另一实施例中,互聚物是乙烯/c6α-烯烃共聚物。在一个实施例中,乙烯/c3-c6α-烯烃互聚物的结晶度百分比≤40%,或≤35%,或≤30%。在另一实施例中,互聚物是乙烯/c3α-烯烃互聚物。在一个实施例中,乙烯/α-烯烃互聚物的结晶度百分比≥20%,或≥22%,或≥24%。在另一实施例中,互聚物是乙烯/c3α-烯烃互聚物,或共聚物。在一个实施例中,乙烯/c3-c6α-烯烃互聚物的分子量分布(mw/mn)≥1.8,或≥2.0,或≥2.1,或≥2.2。在另一实施例中,α-烯烃是c3或c6α-烯烃。在另一实施例中,互聚物是乙烯/c6α-烯烃共聚物。在另一实施例中,互聚物是乙烯/c3α-烯烃互聚物,或共聚物。在一个实施例中,乙烯/c3-c6α-烯烃互聚物的分子量分布(mw/mn)≤3.5,或≤3.2,或≤3.0,或≤2.9,或≤2.8,或≤2.7,或≤2.6。在另一实施例中,α-烯烃是c3或c6α-烯烃。在另一实施例中,互聚物是乙烯/c6α-烯烃共聚物。在另一实施例中,互聚物是乙烯/c3α-烯烃互聚物,或共聚物。在一个实施例中,乙烯/c3-c6α-烯烃互聚物的分子量分布(mw/mn)≥2.0,或≥2.1,或≥2.2。在另一实施例中,互聚物是乙烯/c6α-烯烃共聚物。在一个实施例中,乙烯/α-烯烃互聚物的分子量分布(mw/mn)≤3.0,或≤2.8,或≤2.6。在另一实施例中,互聚物是乙烯/c6α-烯烃共聚物。在一个实施例中,乙烯/c3-c6α-烯烃互聚物的分子量分布(mw/mn)≥2.0,或≥2.1,或≥2.2。在另一实施例中,互聚物是乙烯/c3α-烯烃互聚物,或共聚物。在一个实施例中,乙烯/α-烯烃互聚物的分子量分布(mw/mn)≤3.0,或≤2.8,或≤2.5。在另一实施例中,互聚物是乙烯/c3α-烯烃互聚物,或共聚物。在一个实施例中,乙烯/c3-c6α-烯烃互聚物的重量平均分子量(mw)≤40,000g/mol,或≤35,000g/mol,或≤30,000g/mol,或≤25,000g/mol。在另一实施例中,α-烯烃是c3或c6α-烯烃。在另一实施例中,互聚物是乙烯/c6α-烯烃共聚物。在另一实施例中,互聚物是乙烯/c3α-烯烃互聚物,或共聚物。在一个实施例中,乙烯/c3-c6α-烯烃互聚物的重量平均分子量(mw)≥10,000g/mol,或≥12,000g/mol,或≥15,000g/mol。在另一实施例中,α-烯烃是c3或c6α-烯烃。在另一实施例中,互聚物是乙烯/c6α-烯烃共聚物。在另一实施例中,互聚物是乙烯/c3α-烯烃互聚物,或共聚物。在一个实施例中,乙烯/c3-c6α-烯烃互聚物的数目平均分子量(mn)≤20,000g/mol,或≤18,000g/mol,或≤16,000g/mol,或≤14,000g/mol,或≤12,000g/mol,或≤10,000g/mol。在另一实施例中,α-烯烃是c3或c6α-烯烃。在另一实施例中,互聚物是乙烯/c6α-烯烃共聚物。在另一实施例中,互聚物是乙烯/c3α-烯烃互聚物,或共聚物。在一个实施例中,乙烯/c3-c6α-烯烃互聚物的数目平均分子量(mn)≥4,500g/mol,或≥5,000g/mol,或≥5,500g/mol,或≥6,000g/mol,或≥6,500g/mol,或≥7,000g/mol。在另一实施例中,α-烯烃是c3或c6α-烯烃。在另一实施例中,互聚物是乙烯/c6α-烯烃共聚物。在另一实施例中,互聚物是乙烯/c3α-烯烃互聚物,或共聚物。关于乙烯/c3-c6α-烯烃互聚物,优选α-烯烃包括丙烯、1-丁烯、1-戊烯和1-己烯,并且进一步是丙烯、1-丁烯和1-己烯,并且进一步是丙烯和己烯。在一个实施例中,α-烯烃是c6α-烯烃或c3α-烯烃。在一个实施例中,以互聚物的重量计,乙烯/c3-c6α-烯烃互聚物包含12到40重量%,或14到38重量%,或16到36重量%,或18到34重量%的呈聚合形式的c3-c6α-烯烃。在另一实施例中,α-烯烃是c3或c6α-烯烃。在另一实施例中,互聚物是乙烯/c6α-烯烃共聚物。在另一实施例中,互聚物是乙烯/c3α-烯烃互聚物,或共聚物。在一个实施例中,乙烯/c3-c6α-烯烃互聚物是乙烯/c3-c6α-烯烃共聚物。优选α-烯烃描述于上文中。在一个实施例中,乙烯/c3-c6α-烯烃互聚物的熔融指数(i2或mi),或所计算的熔融指数(i2或mi)大于或等于400g/10min,进一步大于或等于600g/10min,并且更进一步大于或等于800g/10min。在另一实施例中,乙烯/c3-c6α-烯烃互聚物是乙烯/c3-c6α-烯烃共聚物。优选α-烯烃描述于上文中。在另一实施例中,互聚物是乙烯/c6α-烯烃共聚物。在另一实施例中,互聚物是乙烯/c3α-烯烃互聚物,或共聚物。在一个实施例中,乙烯/c3-c6α-烯烃互聚物的熔融指数(i2或mi),或所计算的熔融指数(i2或mi)小于或等于2000g/10min,进一步小于或等于1500g/10min,并且进一步小于或等于1200g/10min。在另一实施例中,乙烯/c3-c6α-烯烃互聚物是乙烯/c3-c6α-烯烃共聚物。优选α-烯烃描述于上文中。在另一实施例中,互聚物是乙烯/c6α-烯烃共聚物。在另一实施例中,互聚物是乙烯/c3α-烯烃互聚物,或共聚物。在一个实施例中,组合物进一步包含至少一种增粘剂和至少一种油。在一个实施例中,以组合物的重量计,组合物包含10到50重量%,或15到45重量%,或20到40重量%的增粘剂。在一个实施例中,以组合物的重量计,组合物包含10到45重量%,或15到40重量%,或20到30重量%的油。乙烯/c3-c6α-烯烃互聚物可以包含如本文中所述的两个或更多个实施例的组合。乙烯/c3-c6α-烯烃共聚物可以包含如本文中所述的两个或更多个实施例的组合。组合物可以包含本文所述的两个或更多个实施例的组合。还提供一种制品,其包含至少一个由本发明组合物形成的组件。在一个实施例中,所述制品进一步包含衬底。在另一实施例中,衬底是选自由以下组成的组:经涂布的衬底、由再循环纸制成的衬底以及其组合。制品可以包含如本文所述的两个或更多个实施例的组合。在一个实施例中,乙烯/c3-c6α-烯烃互聚物在选自以下的至少一个反应器中聚合:再循环环管反应器、连续搅拌槽反应器、活塞流反应器或其组合。在一个实施例中,乙烯/c3-c6α-烯烃互聚物在选自多价芳氧基醚的金属(例如铪、锆或钛)络合物的催化剂存在下聚合。乙烯/c3-c6α-烯烃互聚物在一个实施例中,乙烯/c3-c6α-烯烃互聚物是均匀分支链的线性互聚物,并且进一步是共聚物,或均匀分支链的基本上线性互聚物,并且进一步是共聚物。在一个实施例中,乙烯/c3-c6α-烯烃互聚物是均匀分支链的线性互聚物,并且进一步是共聚物。在一个实施例中,乙烯/c3-c6α-烯烃互聚物是均匀分支链的基本上线性互聚物,并且进一步是共聚物。术语“均匀”和“均匀分支链”用于指乙烯/α-烯烃互聚物,其中α-烯烃共聚单体随机分布在既定聚合物分子内,并且所有聚合物分子具有相同或基本上相同的共聚单体与乙烯的比率。均匀分支链线性乙烯互聚物是乙烯聚合物,其缺乏长链分支,但确实具有短链分支,其来源于聚合到互聚物中的共聚单体,并且其在相同聚合物链内和在不同聚合物链之间都是均匀分布的。这些乙烯/α-烯烃互聚物具有线性聚合物主链、不可测量的长链分支和窄分子量分布。这类聚合物例如由elston在美国专利案第3,645,992号中公开,并且如所显示的,已经在以下文献中研发出了用于使用双-茂金属催化剂制造这类聚合物的后续方法:例如ep0129368;ep0260999;美国专利案第4,701,432号;美国专利案第4,937,301号;美国专利案第4,935,397号;美国专利案第5,055,438号;以及wo90/07526;其各自以引用的方式并入本文中。如所讨论,均匀分支链线性乙烯互聚物缺乏长链支化,正如线性低密度聚乙烯聚合物或线性高密度聚乙烯聚合物的情况一样。均匀分支链线性乙烯/α-烯烃互聚物的商业实例包括来自mitsuichemicalcompany的tafmer聚合物和来自exxonmobilchemicalcompany的exact和exceed聚合物。均匀分支链的基本上线性乙烯/α-烯烃互聚物描述于美国专利案第5,272,236号;第5,278,272号;第6,054,544号;第6,335,410号;和第6,723,810号中;其各自以引用的方式并入本文中。基本上线性乙烯/α-烯烃互聚物具有长链分支。长链分支具有与聚合物主链相同的共聚单体分布,并且可以具有与聚合物主链的长度大致相同的长度。“基本上线性”通常指平均经“每1000个总碳0.01个长链分支”到“每1000个总碳3个长链分支”取代的聚合物。长链分支的长度长于由将一种共聚单体并入聚合物主链中所形成的短链分支的碳长度。一些聚合物可以经“每1000个总碳0.01个长链分支到每1000个总碳3个长链分支”,进一步“每1000个总碳0.01个长链分支到每1000个总碳2个长链分支”,并且进一步“每1000个总碳0.01个长链分支到每1000个总碳1个长链分支”取代。基本上线性乙烯/α-烯烃互聚物形成独特的一类均匀分支链乙烯聚合物。其显著不同于如上文所讨论的众所周知类别的常规均匀分支链线性乙烯/α-烯烃互聚物,并且此外,其与常规的非均匀“齐格勒-纳塔催化剂聚合(ziegler-nattacatalystpolymerized)”线性乙烯聚合物(例如,使用例如anderson等人在美国专利案4,076,698中公开的技术制造的超低密度聚乙烯(uldpe)、线性低密度聚乙烯(lldpe)或高密度聚乙烯(hdpe))不属于同一类别;其与高压、自由基引发的高度分支的聚乙烯(例如低密度聚乙烯(ldpe)、乙烯-丙烯酸(eaa)共聚物以及乙烯乙酸乙烯酯(eva)共聚物)也不属于同一类别。适用于本文中的均匀分支链的基本上线性乙烯/α-烯烃互聚物具有极佳的可加工性,即使其具有相对窄的分子量分布。出人意料地,基本上线性乙烯互聚物的根据astmd1238的熔体流动比率(i10/i2)可以大幅变化,并且基本上与分子量分布(mw/mn或mwd)无关。这些意外特性与常规均匀分支链线性乙烯互聚物(如例如由elston在u.s.3,645,992中所描述)和非均匀分支链的常规“齐格勒-纳塔聚合”线性聚乙烯互聚物(如例如由anderson等人在u.s.4,076,698中所描述)相反与实质上线性乙烯互聚物不同,线性乙烯互聚物(无论均匀或非均匀分支链)具有流变学特性,因此随着分子量分布增加,i10/i2值也增加。长链分支可以使用13c核磁共振(nmr)光谱学测定,并且可以使用randall的方法(《高分子化学物理学综述(rev.macromol.chem.phys.)》,c29(2和3),1989,第285-297页)定量,其公开内容以引用的方式并入本文中。两种其它方法是与低角度激光散射检测器耦合的凝胶渗透色谱法(gpclalls)和与差示粘度计检测器耦合的凝胶渗透色谱法(gpc-dv)。这些长链分支检测技术的使用和基础理论已经在文献中列有充分证据。参见例如zimm,b.h.和stockmayer,w.h.,《化学物理学杂志(j.chem.phys.)》,17,1301(1949),和rudin,a.,《现代聚合物表征方法(modernmethodsofpolymercharacterization)》,johnwiley&sons,newyork(1991)第103-112页。相比于“基本上线性乙烯聚合物”,“线性乙烯聚合物”是指不具有可测量或明显的长链分支的聚合物,即,聚合物经每1000个碳平均小于0.01个长链分支取代。乙烯/c3-c6α-烯烃互聚物可以包含如本文中所述的两个或更多个实施例的组合。乙烯/c3-c6α-烯烃共聚物可以包含如本文中所述的两个或更多个实施例的组合。添加剂和应用通常,聚合物和树脂用一种或多种稳定剂处理,例如抗氧化剂,如irganox1010、irganox1076和irgafos168,各自由basf供应。聚合物通常在挤压或其它熔融过程之前用一种或多种稳定剂处理。其它聚合添加剂包括(但不限于)紫外光吸收剂、抗静电剂、颜料和染料、成核剂、填充剂、滑爽剂、阻燃剂、塑化剂、加工助剂、润滑剂、稳定剂、烟尘抑制剂、粘度控制剂和防结块剂。本发明组合物还可以含有一种或多种热塑性聚合物。本发明的组合物也可以用于多种应用,包括(但不限于)粘着剂、汽车应用、图形艺术、非编织品、面板组合件、高性能胶带、接触热熔胶、纸板涂层、油墨、个人护理和化妆产品、密封剂、色母料和添加剂浓缩物、地毯胶带粘着剂、木工用胶和型材包覆粘着剂。本发明的组合物可以进一步包含增粘剂。例示性增粘树脂包括(但不限于)脂肪族烃、环脂族烃以及芳香族烃和经改性的烃以及氢化形式;萜类和经改性的萜类以及氢化形式;和松香和松香衍生物以及氢化形式;和其混合物。合适的增粘剂包括(但不限于)eastotach100和eastotach115,其皆可以从eastmanchemical购得。在一个实施例中,以组合物的重量计,组合物包含10到60重量百分比,进一步20到50重量百分比,并且进一步30到40重量百分比的增粘剂。在另一实施例中,增粘剂是烃,并且进一步是氢化烃。本发明的组合物可以进一步包含蜡。蜡包括(但不限于)石蜡、微晶蜡、高密度低分子量聚乙烯蜡、聚丙烯蜡、热降解蜡、副产物聚乙烯蜡、费托蜡(fischer-tropschwax)、氧化费托蜡和官能化蜡,如羟基硬脂酰胺蜡和脂肪酰胺蜡。所属领域中通常使用术语“合成高熔点蜡”来包括高密度低分子量聚乙烯蜡、副产物聚乙烯蜡和费托蜡。其它蜡还包括美国专利案第6,335,410号;第6,054,544号和第6,723,810号中描述的蜡;其皆以引用的方式并入本文中。优选蜡包括(但不限于)sasol蜡(例如来自sasolwaxcompany的sasolwaxh1)和费托蜡。在一个实施例中,以组合物的重量计,组合物包含10到60重量百分比,进一步15到50重量百分比,并且进一步20到40重量百分比,并且进一步25到30重量百分比的蜡。本发明组合物可以进一步包含油。油通常用于降低粘着剂的粘度。当使用时,以组合物的重量计,油将通常以小于50,优选小于40,并且更优选小于35重量百分比的量存在。以组合物的重量计,油可以大于或等于2重量百分比,进一步大于或等于5重量百分比,并且进一步大于或等于10重量百分比的量存在。例示性类别的油包括(但不限于)白矿油(如可以从witco购得的kaydol油),和shellflex371环烷油(可以从shelloilcompany购得)和calsol5550(来自calumetlubricants的环烷油)。定义除非相反说明,否则所有测试方法都是截至本发明申请日的现用方法。如本文所用,术语“组合物”包括构成所述组合物的材料混合物以及由所述组合物的材料形成的反应产物和分解产物。如本文中所使用术语“聚合物”是指通过聚合相同或不同类型单体制备的聚合化合物。因此,通用术语聚合物涵盖如下文所定义的术语均聚物(用于指仅由一种类型的单体制备的聚合物,并且应理解,微量杂质可以并入到聚合物结构中)和术语互聚物。微量杂质(例如催化剂残余物)可以并入聚合物中和/或聚合物内。如本文中所使用的术语“互聚物”是指通过聚合至少两种不同类型的单体而制备的聚合物。因此,通用术语互聚物包括共聚物(用于指由两种不同类型的单体制备的聚合物),和由超过两种的不同类型的单体制备的聚合物。如本文中所使用,术语“烯烃类聚合物”是指包含呈聚合形式的50重量%或大量烯烃单体,例如乙烯或丙烯(以聚合物的重量计)并且任选地可以包含一种或多种共聚单体的聚合物。如本文中所使用,术语“丙烯类聚合物”是指包含呈聚合形式的大量丙烯单体(以聚合物的重量计)并且任选地可以包含一种或多种共聚单体的聚合物。如本文中所使用,术语“乙烯类聚合物”是指包含呈聚合形式的≥50重量%,并且优选地,大量乙烯单体(以聚合物的重量计)并且任选地可以包含一种或多种共聚单体的聚合物。如本文中所使用,术语“乙烯/α-烯烃互聚物”是指包含呈聚合形式的≥50重量%,并且优选地,大量乙烯单体(以聚合物的重量计)和至少一种α-烯烃的互聚物。如本文中所使用,术语“乙烯/α-烯烃共聚物”是指包含呈聚合形式的≥50重量%,并且优选地,大量乙烯单体(以共聚物的重量计)和α-烯烃(作为仅有的两种单体类型)的共聚物。如本文中所使用,关于聚合物,术语“集结粒形式”通常指由粒化方法形成的小型聚合物颗粒。这一术语还包括通过将聚合物粉末压缩或粘合成颗粒状集结粒而形成的聚合物颗粒。在一个实施例中,聚合物颗粒是由粒化方法形成。术语“包含”、“包括”、“具有”和其衍生物不意图排除存在任何其它组分、步骤或程序,无论其是否具体公开。为了避免任何疑问,除非另有说明,否则通过使用术语“包含”所要求的所有组合物可以包括任何其它添加剂、佐剂或化合物,无论是聚合的还是其它的。相比之下,术语“基本上由……组成”从任何随后列举的范围中排除任何其它组分、步骤或程序,除了对可操作性来说不是必不可少的那些以外。术语“由……组成”排除未描绘或列出的任何组分、步骤或程序。测试方法熔融粘度根据astmd3236(177℃,350℉),使用布氏数字粘度计(brookfielddigitalviscometer)(型号dv-iii,版本3)和抛弃式铝样品腔室测量熔融粘度。转子为sc-31热熔转子,适用于测量在10到100,000厘泊范围内的粘度。将样品倒入腔室中,然后将腔室插入布氏加热器(brookfieldthermosel)中,并固定就位。样品腔室在底部具有与布氏加热器底部适配的槽口,以确保当转子插入并且旋转时腔室不会转动。将样品(约8-10克树脂)加热到所需温度,直到熔融样品在样品腔室顶部以下一英寸。降低粘度计装置,并且将转子浸没于样品腔室中。继续降低,直到粘度计上的支架在加热器上对齐。开启粘度计并且将其设为在某一剪切率下运行,以粘度计的rpm输出计,所述剪切率会引起转矩读数在总转矩容量的40%到60%的范围内。每分钟收集一次读数,进行15分钟,或者直到所述值稳定为止,此时记录最终读数。熔融指数根据astmd-1238,在190℃/2.16kg条件下测量乙烯类聚合物的熔融指数(i2,或mi)。对于高i2聚合物(i2大于或等于200g/mol),优选由brookfield粘度计来计算熔融指数,如美国专利案第6,335,410号;第6,054,544号;第6,723,810号中所描述。i2(190℃/2.16kg)=3.6126[10(log(η)-6.6928)/-1.1363]-9.3185l,其中η=在350℉下的熔融粘度(cp)。差示扫描热量测定(dsc)差示扫描量热法(dsc)可用于在宽温度范围的下测量聚合物的熔融、结晶和玻璃化转变行为。举例来说,使用配备有rcs(冷藏冷却系统)和自动取样器的tainstrumentsq1000dsc进行这一分析。在测试期间,使用50ml/min的氮吹扫气流。称取约5mg到8mg聚合物样品且置放于dsc盘中,并且将所述盘用钳口盖封闭。随后进行分析,以测定其热特性。通过使样品温度匀变上升和下降以产生热流与温度分布曲线来测定样品的热行为。首先,将样品快速加热到180℃,并且保持等温三分钟,以便去除其热历程。接着,使样品以10℃/分钟的冷却率冷却到-80℃,并且在-80℃下保持等温三分钟。接着,以10℃/分钟加热速率将样品加热到180℃(这是“第二加热”匀变)。记录冷却和第二加热曲线。所测定的值是峰值熔融温度tm和峰值结晶温度tc。使用以下方程式测定熔解热(hf)(焦耳/克)和结晶度百分比(对于聚乙烯样品):结晶度百分比=((hf)/292j/g)×100。熔解热(hf)和峰值熔融温度由第二加热曲线报告。根据冷却曲线测定峰值结晶温度。密度-根据astmd-792测量密度。所测量的密度是“快速密度”,意指所述密度是在成型时间一小时后测定。高共聚单体含量(hcc)方法1使用市售结晶洗脱分级仪器(crystallizationelutionfractionationinstrument;cef)(polymerchar,spain)进行高温热梯度相互相用色谱(tgic)测量(cong等人,《大分子(macromolecules)》,2011,44(8),3062-3072)。使用hypercarb管柱(100×4.6mm,零件号35005-104646,thermoscientific)进行分离。在cef仪器的顶部烘箱中,将填充有27微米玻璃珠粒(目录号gl01918/20-27μm,mo-scispecialtyproducts,llc,rolla,mo,usa)的“8cm×0.48cm(3/16英寸内径)”不锈钢管柱安置在ir侦测器前方。实验参数如下:顶部烘箱/传输线/针温度是150℃,溶解温度是160℃,溶解搅拌设定值是2,样品负载体积是0.400ml,泵稳定时间是15秒,清洁管柱的泵流动速率是0.500ml/m,管柱负载的泵流动速率是0.300ml/min,稳定温度是150℃,稳定时间(前,在装载到管柱之前)是3.0min,稳定时间(后,在装载到管柱之后)是1.0min,sf(可溶性洗脱份)时间是3.0min,从150℃到30℃的冷却速率是3.00℃/min,在冷却过程期间的流动速率是0.00ml/min,从30℃到160℃的加热速率是2.00℃/min,在160℃下的等温时间是10min,洗脱流动速率是0.500ml/min,并且注射环尺寸是140微升。在邻二氯苯(odcb,99%无水级,sigma-aldrich)中,在4.0mg/ml的浓度下,用polymerchar自动取样器在160℃下历时60分钟制备样品。在使用前,在真空烘箱中,在160℃下干燥硅胶40(粒度0.2-0.5mm,目录号10181-3,emd)约两小时。向两升odcb中添加2,5-二-第三丁基-4-甲苯酚(1.6克,bht,目录号b1378-500g,sigma-aldrich)和硅胶(5.0克)。这种“含有bht和硅胶的odcb”现在被称为“odcb”。在使用前,这种odcb用干燥的氮气(n2)鼓泡一小时。用polymerchar(spain)“gpcone”软件平台处理tgic数据。在填充有7.0mlodcb的10ml小瓶中,用以下的混合物进行温度校准:约4到6mgeicosane、14.0mg等规均聚物聚丙烯ipp(分子量mw报告为聚乙烯当量是150,000到190,000g/mol,并且多分散性(mw/mn)是3.6到4.0)和14.0mg均聚物聚乙烯hdpe(零共聚单体含量,mw报告为聚乙烯当量是115,000到125,000g/mol,并且多分散性是2.5到2.8)。在160℃下,溶解时间是两小时。校准过程(对于eicosane洗脱和hdpe洗脱,30℃到150℃)由以下步骤组成:根据加热速率,外推在洗脱期间每个等温步骤的洗脱温度。计算延迟体积。使温度(x轴)相对于ir测量通道色谱图(y轴)偏移,使得eicosane峰值(y轴)与在30.0℃下的洗脱温度重合。按以下方式计算延迟体积:温差(30℃-eicosane峰值的实际洗脱温度)除以所述方法的加热速率,并且接着乘以洗脱流动速率。用这一相同迟缓体积调节来调节每个记录的洗脱温度。线性按比例缩放加热速率,使得hdpe参考物的洗脱峰值温度是150.0℃,同时保持eicosane洗脱峰值温度是30.0℃。观测到聚丙烯的峰值温度是119.3-120.2℃,其是校准方法的验证。用于tgic的聚合物样品的数据处理在下文中描述。在与聚合物样品相同的实验条件下操作溶剂空白(纯溶剂注射)。聚合物样品的数据处理包括以下:a)减去每个检测器通道的溶剂空白,b)温度外推,如校准过程中所描述,c)用由校准过程测定的延迟体积进行温度补偿,和d)在洗脱温度轴中针对30℃和160℃范围进行调节,如由校准的加热速率所计算。用polymerchar“gpcone”软件对色谱图进行积分。当峰值在较高洗脱温度下降低到平坦基线(大致扣除空白色谱图中的零值)和在可溶性洗脱份(sf)的高温侧上的检测器信号的最小或平坦区域时,直线基线绘制出可见差异。当峰值降低到平坦基线区域(扣除空白色谱图中的大致零值)时,基于可见差异建立温度积分上限。基于基线与色谱图的交点(包括可溶性洗脱份)建立温度积分下限。“高共聚单体含量(hcc)”定义为在温度≤65.0℃下洗脱的物质的重量百分比。通过以下方式计算hcc:在小于和包括65.0℃的温度下将ir测量通道(ir色谱图)积分,并且将这个值除以ir测量通道的总积分值。图4是用于测定比较性共聚物的hcc值的实例色谱图。高共聚单体含量(hcc)方法2ht-tgic(或tgic)测量利用市售结晶洗脱分级仪器(cef)(polymerchar,spain)进行高温热梯度相互相用色谱(ht-tgic,或tgic)测量(cong等人,《大分子》,2011,44(8),3062-3072)。方法2使用配备有ir-4检测器的cef仪器(如由polymerchar,spain市售),和低孔隙度石墨作为衬底或固定相。将低孔隙度石墨填充到尺寸是0.46(id)×25(长度)cm的管柱中。在cef仪器的顶部烘箱中,将填充有27微米玻璃珠粒(目录号gl01918/20-27μm,mo-scispecialtyproducts,llc,rolla,mo,usa)的“8cm×0.48cm(3/16英寸内径)”不锈钢管柱安置在ir侦测器前方。使用polymerchar仪器控制软件。polymerchar仪器控制软件的实验参数是:顶部烘箱/传输线/针温度是150℃,溶解温度是160℃,溶解搅拌设定值是2,泵稳定时间是15秒,清洁管柱的泵流动速率是0.500ml/m,管柱负载的泵流动速率是0.300ml/min,稳定温度是150℃,稳定时间(前,在装载到管柱之前)是3.0min,稳定时间(后,在装载到管柱之后)是1.0min,sf(可溶性洗脱份)时间是5.0min,从150℃到30℃的冷却速率是3.00℃/min。在冷却期间的流动速率是0.04ml/min,并且从30℃到150℃的加热速率是2.00℃/min,并且在150℃下的等温时间是10分钟。洗脱流动速率是0.500ml/min,并且注射环尺寸是200微升。根据石墨管柱的长度调节在冷却过程期间的流动速率,其中在冷却循环结束时,所有聚合物洗脱份必须保留在管柱上。在160℃下,在odcb(下文中定义)中4.0mg/ml的浓度下,用polymerchar自动取样器历时60分钟制备样品。在使用前,在真空烘箱中,在160℃下干燥硅胶40(粒度0.2-0.5mm,目录号10181-3,emd)约两小时。将2,6-二-叔丁基-4-甲苯酚(1.6克,bht,目录号b1378-500g,sigma-aldrich)和硅胶40(5.0克)添加到两升的邻-二氯苯(odcb,99%无水级,sigma-aldrich)中。这种“含有bht和硅胶的odcb”被称为“odcb”。在使用前,这种odcb用干燥的氮气(n2)鼓泡一小时。用polymerchar(spain)“gpcone”软件平台处理tgic数据。在填充有7.0mlodcb的10ml小瓶中,用约4到6mgeicosane、14.0mg等规均聚物聚丙烯ipp(多分散性是3.6到4.0,并且分子量mw报告为聚乙烯当量是150,000到190,000,并且多分散性(mw/mn)是3.6到4.0,dsc熔融温度是158-159℃,用下文指定的方法)和14.0mg均聚物聚乙烯hdpe(零共聚单体含量,mw报告为聚乙烯当量是115,000到125,000,并且多分散性是2.5到2.8)。在160℃下,溶解时间是两小时。校准过程(对于eicosane洗脱和hdpe洗脱,30℃到150℃)由以下步骤组成:根据加热速率外推在洗脱期间的每个等温步骤的洗脱温度(说明于图5中(用于tgic温度校准的洗脱温度的外推))。计算延迟体积。使温度(x轴)相对于ir测量通道色谱图(y轴)偏移,使得eicosane峰值(y轴)与在30.0℃下的洗脱温度重合。通过温差(30℃-eicosane最大峰值的实际洗脱温度)除以方法的加热速率并且然后乘以洗脱流动速率来计算延迟体积。用这一相同延迟体积调节来调节每个记录的洗脱温度。以线性方式标度加热速率,使得观察到的hdpe参考值具有150.0℃的洗脱峰值最大温度,而eicosane洗脱峰值最大温度保持在30.0℃下。观察到聚丙烯的峰值温度在119.3℃-120.2℃范围内,其是校准方法的验证。用于tgic的聚合物样品的数据处理在下文中描述。在与聚合物样品相同的实验条件下操作溶剂空白(纯溶剂注射)。对聚合物样品的数据处理包括:对每个检测器通道的溶剂空白的扣除、如校准方法中所述的温度外推、用由校准方法测定的延迟体积进行的温度补偿以及将洗脱温度轴调节到30℃和150℃范围,如由校准的加热速率计算。色谱图(ir-4检测器的测量通道)用polymerchar“gpcone”软件进行积分。当峰值在较高洗脱温度下降低到平坦基线(大致扣除空白色谱图中的零值)和在可溶性洗脱份(sf)的低温侧上的检测器信号的最小或平坦区域时,直线基线绘制出可见差异。当峰值降低到平坦基线区域(扣除空白色谱图中的大致零值)时,基于可见差异建立温度积分上限。基于基线与包括可溶性洗脱份的色谱图的交点建立温度积分下限。方法2的hcc定义为在温度积分下限与59.0℃之间洗脱的聚合物洗脱份-参见以下方程式h2。对于方法2,通过将28.2℃到59.0℃的面积除以总面积(28.2℃到160℃)并且将这个商乘以100来测定hcc值。还参见图6,其显示各自通过hcc方法2获得的聚合物eh-2和eh-7的ht-tgic色谱图。在±0.1或更小的标准差内,使用本文中所描述的方法(1和2)获得的hcc值提供相同的值。管柱制备-低孔隙度固定相-hcc方法2低孔隙度石墨,球形颗粒,其中d50是20-24微米,d10大于12微米,并且d90小于42微米。孔隙度特征列举于表a中。将低孔隙度石墨填充到0.46(内径)×25(长度)cm的管柱中。表a:孔径分布和表面积表征**使用直径是0.003到1微米的孔。用于填充方法2的管柱的硬件-ht-tgic从agilenttechnologies(以前是polymerlabinc.)获得不锈钢管柱、玻璃料、管柱的末端配件。使用agilent1100型液相色谱泵进行浆料填充方法。tcb(1,2,4-三氯-苯)是浆料介质。浆料填充储集器由具有valco末端配件的“0.46cm”内径不锈钢管构成。储集器的长度是150mm。使用标准1/4"外径管接头连接填充储集器与空的分析管柱。用于填充管柱的方法填充的管柱呈现良好的质量转移特性,包括在标准流动速率和温度操作条件下的低背压、对突然变化条件引起的振动的低敏感性以及不具有通道和空隙空间。填充的管柱具有充足的内部液体体积以允许研究动态冷却对组分解析的作用。动态冷却是在cef和ht-tgic的冷却过程期间使用缓慢流动速率的方法(monrabal等人,《高分子专题研讨会(macromol.symp.)》257,71-79(2007),和cong等人,《高分子学(macromolecules)》,11,44(8),3062))。制备低孔隙度管柱的方法首先利用(1)通过使用分接头填充(tap-and-fill)方法进行干燥填充,其中通过对管柱进行分接或使用电振动工具来安置所添加的材料,接着(2)浆料填充方法,其使用底物的悬浮液或浆料,其中将浆料在流动条件下泵送到管柱中(striegel,yau等人,《现代尺寸排阻液相色谱法(modernsizeexclusionliquidchromatography)》,wiley,第2版本,第6章)。对于简单的分接头填充方法,管柱是垂直悬浮的。通过漏斗以小型递增形式添加衬底,同时对管柱进行分接和振动以使衬底沉降。当衬底与管柱的末端齐平时,添加末端配件,并且密封管柱。在使用前的标准操作是调节管柱,并且检查用于沉降的床或空隙。如果发现存在空隙,那么添加更多的填充物以与管柱的末端齐平。对于浆料填充方法,将衬底材料干式添加到空的管柱中。接着组装储集器和管柱与末端配件并且连接到agilent泵。以1ml/min的流动速率向上泵送tcb使其通过储集器,直到从管柱置换空气。短暂地停止流动,接着将管柱和储集器倒置成下流式位置。以3-5ml/min泵送tcb使其通过管柱保持至少二十分钟,或直到系统压力达到2500psig。使管柱与填充储集器断开连接,并且用平坦的叶片式刮板去除管柱末端的任何过量填充物,以提供与导管末端的齐平。将末端配件就地紧固,并且使管柱准备进行调节。管柱调节将新填充的管柱安置在ht-tgic仪器中,并且在室温下建立0.1ml/min的流动速率。取决于材料和其填充有效性,此时的背压通常是2-10巴。在各步骤中,流动速率增加0.1ml/min,使得压力在每次增加之间稳定,直到0.7或1.0ml/min。管柱温度增加到60℃,并且接着在流动下,使用线形温度匀变以10℃/min将管柱加热到140℃。保持这一最终温度20分钟,并且接着使管柱以10℃/min冷却到100℃,并且准备用于测试。用于孔径分布和孔隙度的汞压孔率测定法通过汞压孔率测定法获得孔径分布。用可以从micromeritics获得的micromeriticsautoporeiv9520进行汞压孔率测定法分析。样品在110℃下干燥2小时,并且接着在分析之前,在真空中以机械方式除气以从样品表面移除任何以物理方式吸附的物质(即,水分)。测定条件包括hg填充压力是0.50psia,hg接触角是130°,hg表面张力是485dyn/cm,hg密度是13.53g/ml,抽真空时间是30分钟,大孔穿透计(粉末类型:1.131茎干体积)具有5cc球状物,平衡时间是30秒,92点压力表(75个入侵加17个挤压压力点)并且机械抽真空<50μmhg。在约39psia(4.6μm)下收集从低到高压力交叉点。生成所用的压力表是为了在0.5到60,000psia的对数标尺上使压力均匀递增分布,并且用于检测直径是0.003到400μm的孔径。随着压力从真空逐渐递增到最大值约60,000psia,迫使汞进入越来越小的孔中。为了验证仪器正确工作,分析二氧化硅-氧化铝参考材料(micromeritics批号a-501-46)。参考样品的所报告的中值孔径(体积)是0.0072±0.0005μm。参考材料的autopore报告的中值孔径(体积)是0.0071μm。通过使用配备有micromeriticsautoporeiv9520的数据处理软件排除粒子间入侵来计算孔隙度。在从假设由材料占据的体积排除所有大于约0.003μm的孔的体积之后计算骨架密度。氮气吸附/解吸附(b.e.t.)用micromeriticsacceleratedsurfacearea&porosimetry仪器(asap2405)进行氮气吸附/解吸附分析。在分析之前,样品在真空中,在200℃下除气约24小时。将约0.5克“按原样接收的”样品用于分析。通常,在精确度<3%rsd(相对标准偏差)下获得b.e.t.表面积。仪器使用静态(体积)样品供料方法,并且测量可以在液氮温度下以物理方式吸附在固体上的气体(氮气)量。对于多点b.e.t.测量,在恒定温度下,在预先选择的相对压力点处测量氮气吸收体积。相对压力是在77k的分析温度下所施加的氮气压力与氮气的蒸气压的比率。通过这种方法检测约17到3,000埃直径的孔径。用于氮气吸附/解吸附等温线的测定条件包括15秒平衡区间,97点压力表(40个吸附点、40个解吸附点、多点b.e.t.表面积、20个微孔点和1点式总孔隙体积)、5%/5mmhgp/po容许度和120minpo区间。使用由micromeriticsacceleratedsurfacearea&porosimetry仪器(asap2405)配备的数据处理软件进行bet计算。粒度分布(d50、d10、d90)激光衍射粒度分析仪利用fraunhofer光散射理论。基于激光衍射的粒度分析依赖于粒子以某一角度通过激光束散射光,其中散射光强度与粒子尺寸直接相关。使用配备有universalliquidmodule的beckmancoulterls13320激光衍射仪器测定基础粒子的粒度分布。使“0.25克”样品分散于40ml干净并且无气泡的去离子水(di)中。在实际操作之前,仪器进行常规背景调查并且减去任何可能在装载待测量的粒子之前已经存在的粒子。在室温下,用磁性搅拌器搅拌混合物以使粒子均匀分散。向基础粒子的混合物中添加一滴“micro90表面活性剂(产品)”。将数滴稀分散液注射到分析仪的液体模块端口中,直到获得8%暗度。接着将仪器设置成运行模式。每次运行60秒。以一分钟间隔进行三次连续操作,以确保暗度保持恒定,并且粒子不溶解或聚结。使用等效球径表征粒子的尺寸分布。在0.4到2000微米尺寸范围内进行粒度测量。软件报告的统计分析中不包括测量范围外的粒子。用由beckmancoulterls13320仪器提供的软件进行平均粒度的数据获取和计算。d10、d50和d90分别定义为在10%点、50%点和90%点处的累计分布曲线的直径(以体积计)。用beckmancoulter的对照样品进行的正常操作的乳胶标准检验仪器。常规gpc高温凝胶渗透色谱(htgpc)系统由来自agilent的pl-220模型或来自polymerchar的gpcir模型组成。色谱柱和传送室在140℃下操作。使用三个聚合物实验室“10-μm混合b管柱”和1,2,4-三氯苯的溶剂。在“含0.1g聚合物的50ml溶剂”的浓度下制备样品。用于制备样品的溶剂含有“200ppm抗氧化剂bht”。通过在160℃下搅拌/轻微振荡四小时来制备样品。注射体积是200微升,并且流动速率是1.0ml/min。用从agilent购得的二十一个窄分子量分布聚苯乙烯标准进行gpc管柱集合的校准。使用以下方程式1将聚苯乙烯标准峰值分子量转化成聚乙烯分子量:m聚乙烯=a(m聚苯乙烯)b(方程式1),其中m是分子量,a的值是0.4316,并且b等于1.0(参考:t.williams和i.m.ward,《聚合物科学聚合物期刊(j.polym.sci.,polym.let)》,6,621(1968))。确定三阶多项式以建立分子量校准对数随洗脱体积的变化。使用“gpcone”软件(polymerchar)进行聚乙烯当量分子量计算。根据以下方程式2和方程式3计算数目平均和重量平均分子量:在方程式2和3中,wfi是第i个分量的重量分数,并且mi是第i个分量的分子量。重量平均分子量(mw)的精确度<2.6%。可以进行重复分析以实现这一精确度。老化流动性测试方法为了定量集结粒与集结粒粘性,研发漏斗测试。这一测试是以增加粒子间相互相用(粘性)将降低浸渍漏斗的排出率的基本概念为基础。排出率的变化可以与聚合物集结粒的表面特性(即,粘性)的变化相关。测试装置(参见图3)由连接到圆柱(4.15英寸直径)的浸渍玻璃漏斗组成。圆柱形部分提供所需容量,使得可以测试大量集结粒,并且避免区分小型排出时间值的问题。重复测试五次以用于统计目的。对“按原样接收”的样品和在预定储藏温度下调节集结粒之后,历时预定持续时间测量集结粒的排出率。集结粒在42℃下“热处理”或“老化”三周。被调节的集结粒在21℃下冷却隔夜以实现恒定温度。如上文所论述,聚合物(约2500g;集结粒形式;30±10个集结粒/克)在烘箱中,在42℃下热处理三周。从烘箱移出聚合物,并且在21℃下冷却12小时。用聚合物集结粒(2500g)填充漏斗,并且测量用于完成集结粒从漏斗排出所需的时间,并且使用以下方程式计算排出率。排出率或流动性(g/s)=漏斗中集结粒的量(g)/排出所需时间(s)流动性是集结粒粘性的指标。已测定120g/s的流动性是实现聚合物集结粒的可接受操作特征所需的最小流动速率。然而,优选甚至更高的速率以便更好地操作聚合物集结粒。更高的集结粒流动性值对应于更多的自由流动和更少的粘性集结粒。可以用≤5000ppm或更少的一种或多种稳定剂)(例如irgonox1010、irganox1076和irgafos168)使聚合物稳定;然而,这类少量稳定剂将不会影响聚合物的流动性结果。热应力根据由封装专业协会(instituteofpackagingprofessions;iopp)制定的“《用于测定热熔融粘着剂的热应力耐受性的推荐测试方法(suggestedtestmethodfordeterminingtheheatstressresistanceofhotmeltadhesives)》”,方法t-3006来测量热应力耐受性(热应力)。为了制备一份样品,通过用olingerbondtester,在较短的试样中间施用0.00014lb/in的组合物(0.00028lb)使尺寸是2英寸(50.8mm)×3-3/16in(81mm)和2in(50.8mm)×5-1/2in(139.7mm)的两张纸板型试样(沿长度方向切割有凹槽)粘合。将组合物垂直施用于较短试样的中心的凹槽中并且使试样粘合,使得从较长试样的一端计,组合物是3/4in(19mm)。对于每种粘着剂配制物制得六个复本。样品装载到样品固持器中,使短试样末端与样品固持器边缘对准。样品在宽盘通过翼形螺母固定的情况下保持在适当的位置。在距粘合处3.94in(100mm)处置放“200g砝码”。砝码通过将砝码上的栓钉放入长试样中的孔中来固定。接着,将样品固持器在测试温度下放入对流烘箱中24小时。如果6个粘着处中的至少4个未失效,那么认为样品在测试温度下通过耐热性测试。改变烘箱温度,直到测定最大传递热应力耐受性。对于每种测试温度使用所有新粘着的试样。结果议热应力温度(℃)形式报告。剥离粘着失效温度(paft)和剪切粘着失效温度(saft)使用两片“40磅”牛皮纸制备测试样品,每片的尺寸是6in×12in(152mm×305mm)。在底部纸片上,纵向并且在中心处产生1英寸(25mm)间隙,在2层牛皮纸之间,在1"中心间隔的任一侧上包夹羊皮纸。将粘着剂配制物加热到177℃(350℉),在底部衬底的顶部中心分配约7克熔融粘着剂。接着,在粘着剂可能不当地稠化之前,两个玻璃棒(各自具有0.25"直径),一个棒缠绕有4.5"压敏性条带,如间隔2"间隔的遮蔽带,紧密地架在底部衬底上,接着第二个棒架在顶部衬底和(在两个棒之间)第二张纸上,并且棒沿纸片的长度向下滑动。进行这种操作,使得第一个棒均匀地分散间隙中的粘着剂,并且第二个棒均匀地压缩间隙顶部的第二纸片。因此,在两张纸之间产生粘着键的单一“1英寸(25.4mm)宽”条带。这样粘结的纸张横向切成宽度为1英寸(25.4mm)且长度为3英寸(76.2mm)的条带,每个条带在中心具有1in×1in(25mm×25mm)的粘着剂样品粘结。在测试之前24小时,在室温、23℃和50%相对湿度下调节所有测试条带。接着,将条带用于paft和saft测试。根据astm标准测试d-4498测定剥离粘着失效温度(paft)和剪切粘着失效温度(saft)。使用可编程烘箱。烘箱温度从30℃开始并且以0.5℃/min升高,直到粘着键失效并且记录粘着键未失效的最高温度。对于paft测试,使“100克砝码”连接到一个牛皮纸衬底的一端。将另一张牛皮纸的同一端固定到固定的样品固持器。这一设置与180度剥离作用类似。测试三个测试条带并且报告平均温度。对于saft测试,使“500克砝码”连接到牛皮纸衬底的一端。将另一张牛皮纸的相对端固定到固定的样品固持器。这一设置与180度剪应作用类似。测试三个测试条带并且报告平均温度。纤维撕裂(ft)百分比根据标准化方法测定使用inland波纹状卡板的每种粘着剂配制物的纤维撕裂(ft)百分比。使用olingerbondtester,一种用于形成和撕裂测试粘结的机械测试器件,将一滴粘着剂施用于卡板试样(5cm×6cm)的中心(珠粒以0.00014lb/in,以约0.75英寸股线形式置放)。在粘着剂顶部快速置放第二卡板试样(2.5cm×6.5cm)。轻轻施加指压,持续约3秒,以将粘结固定在适当位置。在室温和50%相对湿度下调节测试样品至少四小时。接着,在测试温度下调节样品五小时到24小时。对于每种测试温度,各自手动拉开五个测试样品,并且测定各粘结区域内的纤维撕裂量(%)记录平均纤维撕裂百分比。开放时间和凝固时间使用olingerbondtester,一种用于形成和撕裂测试粘结的机械测试器件来测定凝固时间和开放时间特性。将olingerbondtester加热到350℃(177℃)以使粘着剂配制物溶融。底部衬底,2.5英寸(63.5mm)×2英寸(50.8mm)波纹板,在粘着剂容器下的轨迹移动,所述粘着剂容器一滴粘着剂,约1/16"(1.6mm)到1/8"(3.2mm)宽和1"(25.4mm)长(粘结测试仪在0.00014lb粘着剂/in下操作)。使粘着剂容器压力升高或降低,以便维持珠粒尺寸一致。在两巴压力下,将顶部衬底,2.5英寸(63.5mm)×2英寸(50.8mm),施用于含有粘着剂条的底部衬底。olinger配备有两个计时器,用于以秒为单位测量潜在凝固时间和开放时间。开放时间是对一个衬底施用粘着剂与粘结第二衬底之间的最长时段,其在室温下,在3个样品中引起至少50%纤维撕裂。对于测定开放时间,压缩时间设置成在室温下实现3个样品的100%纤维撕裂所需的时间。“对一个衬底施用粘着剂与粘结第二衬底之间的时段(t)”初始设置成10秒,并且以10秒间隔增加,直到在室温下实现1个样品的小于50%纤维撕裂。使时间t缩短5秒并且测定纤维撕裂%。最终,使时间t以一秒间隔变化,以测定在室温下实现3个样品的50%或更大纤维撕裂的最大允许时间。凝固时间是在室温下实现3个样品的至少50%纤维撕裂的纤维撕裂粘结所需的最小压缩时间。对于这一测试,开放时间设置成两秒。以在两巴压力下,将顶部衬底压到底部衬底上的形式形成粘结。在预设压缩时间之后,通过在室温下从底部衬底拉开顶部衬底来进行撕裂测试。接着进行目测评估,以测定在预设压缩时间下实现的纤维撕裂百分比。使凝固时间以一秒间隔变化,以测定在室温下实现3个样品的50%纤维撕裂所需的压缩时间,以及在室温下实现小于75%纤维撕裂所需的压缩时间(五个测试样品的平均值)。以秒为单位,以最短时间形式记录凝固时间,在所述凝固时间下,在室温下获得3个样品的50%纤维撕裂的最小值。13cnmr通过在10mmnmr管中,向0.2g样品中添加约2.6g四氯乙烷-d2(0.025m于乙酰基丙酮酸铬(松弛剂)中)来制备样品。通过将管及其内含物加热到150℃来使样品溶解且均匀化。使用jeoleclipse400mhz光谱仪收集数据。在130℃下,使用每个数据文件4000次扫描、6秒脉冲重复延迟、25,200hz光谱宽度和32k数据点文件尺寸获取数据。现将在以下实例中详细描述本公开的一些实施例。实验a.代表性聚合-概述在连续溶液聚合反应器中制备每种聚合物。使所有试剂(单体、共聚单体和氢)溶解于溶剂载剂进料流中并且注射到等效于美国专利案5,977,251中所描述的循环、单环管反应器中。溶剂稀释剂是包含5到9个碳的饱和脂肪族烃分子(isopare)。催化剂是(钛,[n-(1,1-二甲基乙基)-1,1-二甲基-1-[(1,2,3,4,5-η)-2,3,4,5-四甲基-2,4-环戊二烯-1-基]硅烷胺并(2-)-κn][(1,2,3,4-η)-1,3-戊-二烯]-)。使用两种共催化剂:三-(2,3,4,5,6,-五氟苯基)硼烷(共催化剂1)和经改性的甲基铝氧烷(共催化剂2)。在注射之前混合两种共催化剂,并且将这一混合物与催化剂分开地供应到反应器中。调节反应器中催化剂的浓度以控制反应器中的乙烯浓度使其处于目标值。调节两种共催化剂物质中的每一种的浓度以控制每种共催化剂与催化剂物质的摩尔比使其处于目标值,以便完成催化剂复合物的活化。在反应器的进料中,调节α-烯烃共聚单体(1-己烯或丙烯)浓度以控制聚合物的密度,并且调节氢浓度以控制聚合物的熔融粘度(或分子量)。在135℃的反应温度和超过35巴的反应压力下进行每次聚合。在溶剂中稀释催化剂杀死剂(去离子水)和抗氧化剂(irganox1010),并且注射到聚合物料流中,离开反应器以终止反应,并且保护聚合物避免氧化。通过从溶剂、未反应单体、非反应性共聚单体和氢移出聚合物产物的系统来处理反应器流出物。使用水下粒化机将聚合物熔融物转化成固体颗粒。用1,000ppmw的irganox1010使聚合物稳定。聚合条件的概述显示于以下表1a和1b中。聚合物特性列举于以下表2a到2d和表3中。表1a:eh共聚物表1b:ep共聚物参数ep1ep2ep3反应温度(℃)135135135反应压力(巴)363636反应器中的乙烯浓度(kg/m3)15.715.19.0反应器中的丙烯浓度(kg/m3)28.226.220.7反应器中的聚合物浓度(重量%)22.122.333.4氢/产物比率(h2(g)/聚合物(吨))248298215共催化剂-1/催化剂摩尔比4.03.93.0共催化剂-2/催化剂摩尔比6.05.91.7表2a:eh共聚物*hcc方法2**基于对来自相同聚合物制备方法的另一批聚合物进行的流动性测量来评估。表2b:eh共聚物实例mn(g/mol)mw(g/mol)mwdeh18564202422.36eh28972217522.42eh38407191302.28eh47825182832.34eh57866181852.31eh67702181492.36eh77799186752.39表2c:eh共聚物*tm2是“最高峰值熔融温度”,定义为观察到最高热流的温度。如表2a和3中所见,已发现每种聚合物的老化流动性超过130g/s,即使一些聚合物具有更高的hcc值(例如超过6重量%)。还参见图1和2。表2d:eh共聚物实例乙烯重量%*己烯重量%*+/-eh169.730.30.3eh368.931.10.2eh467.532.50.1eh667.632.40.3eh270.229.80.4eh567.432.60.4eh767.532.50.5*13cnmr表3:ep互聚物ep1ep2ep3乙烯重量%*81.580.679.7丙烯重量%*18.519.419.3己烯重量%*1.0密度0.8920.8900.890粘度(177℃)18326778810977tm1(℃)79.978.477.9熔解热(j/g)79.684.876.0结晶度(%)27.229.025.9tc1(℃)66.266.063.5δh结晶度(j/g)85.184.4mn865170418157mw201741620718420mw/mn2.32.32.3hcc***(重量%)01.12.0老化流动性(g/s)>130**>130**212*13cnmr。**基于ep3的老化流动性值以及ep1和ep2的较低hcc值评估老化流动性。***hcc方法2。粘着剂制备通过罐混合器方法进行每种热熔融粘着剂形成。称重总共50g粘着剂组分并且置放于内衬有环氧化物的16oz铝罐中。接着,将罐置放于预先设置成350°f的烘箱中30分钟以使组分熔融。接着,将罐转移到加热块,保持在350°f下,以混合组分。混合装置包含加热板(1000型modelspa1025b,由thermolyne制造)、数字温度控制器(model89000-10,由cole-parmer制造)、机械搅拌器(modell1u08flabmastersimixer,由lightnin制造),并且以上加热块配备有热电偶。使铝罐的内含物平衡约五分钟,接着搅拌。混合速度从0逐渐缓慢上升到100rpm,并且使用氮气垫填充铝罐的顶部空间。继续混合约五分钟。接着,在环境温度下,将罐的内含物(粘着剂)倒在teflon片上并且冷却。一旦粘着剂处于室温下,将其储存在ziploc包中直到使用。在以下测试条件下测试每种粘着剂配制物:在各种温度下的paft、saft、热应力温度、开放时间、凝固时间和纤维撕裂。结果显示于以下表4中。如表4中所见,每种热熔融粘着剂在paft、saft、热应力和纤维撕裂方面显示良好粘着效能,并且各自具有适合的开放时间和凝固时间。表4:粘着剂配制物(重量份)和结果实例f1f2f3f4eh135eh435eh635eh735eastotach11535353535sasolh130303030irganox10100.50.50.50.5测试在177℃下的粘度(cp)461.9337.7899.8477.9paft(℃)59.756.056.755.0saft(℃)96.595.396.393.5热应力温度(℃)60607065在-40℃下的ft(%)98897450在-17℃下的ft(%)93978067在0℃下的ft(%)97998683在23℃下的ft(%)100969487在60℃下的ft(%)84758658开放时间(秒)14151511凝固时间(秒)1212当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1