用于在高温溶液聚合方法中制备聚乙烯共聚物的催化剂体系与流程
本发明涉及一种新的催化剂体系,其能够在高温溶液聚合方法中制备聚乙烯共聚物。该新的催化剂体系包含特定取代的桥连茂铪催化剂络合物以及硼基助催化剂,所述茂铪催化剂络合物包含环戊二烯基(cp)配体,芴基(flu)配体和连接该两个配体的特定共价桥。这种组合显著地使催化剂体系具有高生产率和高的溶解度、分子量能力和共聚单体引入能力。
背景技术:
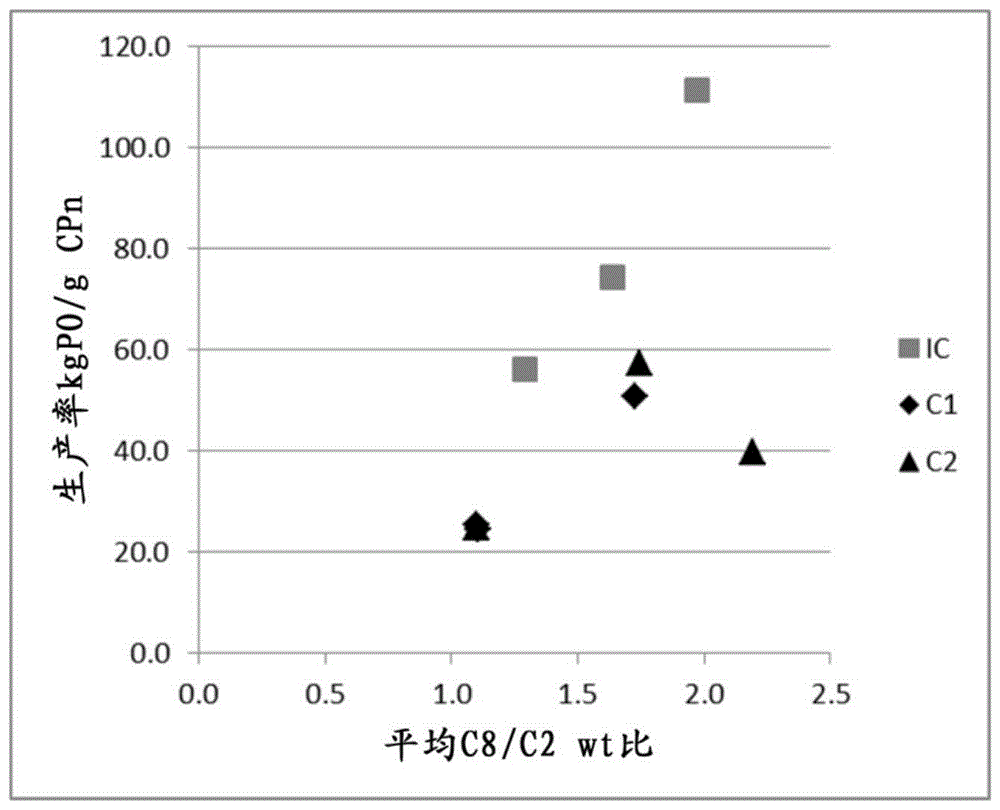
:茂金属催化剂已用于制备聚烯烃多年。无数的学术和专利出版物描述了这些催化剂在烯烃聚合中的用途。现在茂金属已在工业上使用,并且通常使用具有不同取代模式的基于环戊二烯基的催化剂体系来制备聚丙烯以及聚乙烯。这些茂金属催化剂的几种已被描述用于在溶液聚合中使用以制备聚乙烯均聚物或共聚物。例如,wo2000024792描述了一种催化剂体系,其包含衍生自a)双环戊二烯基铪有机金属化合物的茂铪催化剂络合物,其具有i)至少一个未取代的环戊二烯基配体,或芳香稠环取代的环戊二烯基配体且在所述配体上不具有其它的取代基;ii)一个取代的或未取代的,芳香稠环取代的环戊二烯基配体,和iii)连接这两个环戊二烯基配体的共价桥。该桥可以是被两个芳基基团取代的单个碳,这些芳基基团中的每一个均被c1-c20烃基或烃基甲硅烷基基团取代,其中这些取代基中的至少一个是直链c3或更大的取代基。此外,所述催化剂体系包含活化助催化剂,其优选为包含卤代四芳基取代的第13族阴离子的前体离子化合物。ep2511305也涉及桥接的cp-flu茂金属络合物。实施例中所示的c-基桥是取代的亚甲基桥,其中取代基是相同的(例如二甲基,二苯基,二苄基等)或连接在一起形成一个环(例如亚环己基)。yano及其同事(文献1至4)研究了配体结构与多种cp-flu茂金属对高温乙烯均聚和共聚的影响。1.a.yano,m.sone,s.hasegawa,m.sato,a.akimoto,macromol.chem.phys.1999,200,933.2.a.yano,s.hasegawa,t.kaneko,m.sone,m.sato,a.akimoto,macromol.chem.phys.1999,200,1542.3.a.yano,m.sone,s.yamada,s.hasegawa,m.sato,a.akimoto,journalofmolecularcatalysisa:chemical2000,156,133.4.s.hasegawa,m.sone,m.tanabiki,m.sato,a.yano,journalofpolymerscience:parta:polymerchemistry2000,38,4641.这项工作的主要教导在于:i)对于zr络合物和类似物hf络合物这二者,在c2均聚和c2/c6共聚中,与mephc或me2c桥相比,ph2c桥提供了更高的活性和更高的mw。ii)在cp配体上加入取代基不会显著改变催化剂性能iii)以zr为金属,在c2/c6共聚中,在flu配体的2,7位上加入甲基或叔丁基取代基会增加共聚物的分子量,但不会影响共聚单体反应性和催化剂生产率。iv)以hf为金属,在c2/c6共聚中,在flu配体的2,7位上加入叔丁基取代基会稍微提高催化剂生产率,但共聚物的分子量和共聚单体反应性几乎不受影响。v)zr络合物比hf络合物活性高,但ph2c(cp)(flu)zrcl2络合物与其hf类似物相比产生更低的共聚物mw。在上述引用的文献和专利中,没有一个提到高生产率与高的溶解度、分子量能力和共聚单体引入能力相结合的催化剂体系。此外,上述引用的文献和专利没有解决由芳基基团取代但第二个取代基为非芳基取代基的c原子所构成的桥的空间阻碍,在较高聚合温度下对铪络合物的催化剂性能的影响。然而,为使制备乙烯共聚物的方法高效,重要的是所用的催化剂体系满足一系列非常严格的要求。对于较高级共聚单体的共聚单体反应性(共聚单体引入),催化剂分子量能力(对于给定的聚合物密度、单体浓度和聚合温度,最低可达到的熔体指数)和催化剂热稳定性必须确保具有密度低至~0.850g/cm3和熔体指数mi2(190℃,2.16kg)低至~0.3g/10min的共聚物的生产具有高生产率(为了用尽可能用量少的催化剂制备得到最多的聚乙烯)。过渡金属络合物在脂肪烃中的溶解度对于确保该络合物在溶液方法中的最佳使用也是重要的。尽管在茂金属催化剂领域已经做了很多工作,但仍然存在需要找到用于乙烯共聚的新的催化剂体系,其能够生产具有所需性能的聚合物,且其具有高生产率和高的溶解度、分子量能力和共聚单体引入能力。技术实现要素:因此,本发明人着手开发一种新的催化剂体系,其在生产率、溶解度、分子量能力和共聚单体引入能力方面具有优于上述提及的聚合催化剂体系的聚合性能。本发明人现已发现一类新的烯烃聚合催化剂体系,其能够解决上述披露的问题。特别地,本发明将特定茂金属络合物与硼助催化剂结合使用。因此,从一个方面来看,本发明涉及用于在高温溶液方法中制备乙烯共聚物的催化剂体系,该催化剂体系包括(i)式(i)的茂金属络合物其中m是hf或与zr的混合物,条件是式i的络合物按摩尔计超过50%具有m=hfx是σ配体r彼此相同或不同并且可以是饱和的直链或支链c1-c10烷基、c6-c10芳基、c4-c10杂芳基、c6-c20烷基芳基或c6-c20芳基烷基基团,其可以任选地含有至多2个杂原子或硅原子r1是c6-c10芳基或c6-c20烷基芳基基团,任选地含有至多2个杂原子或硅原子或c4-c10杂芳基基团r2是式(ii)的c4-c20环烷基基团,任选地在β-位带有烷基取代基其中r’可以彼此相同或不同,并且可以是氢或被定义为r且n为1-17和(ii)含硼助催化剂。从另一方面来看,本发明提供了一种用于制备乙烯共聚物的方法,其包括在催化剂存在下,在高于100℃的温度下的高温溶液方法中聚合乙烯和c4-10α-烯烃共聚单体,所述催化剂包括:(i)如上定义的式(i)的茂金属络合物和(ii)含硼助催化剂。从另一方面来看,本发明提供一种通过上文定义的方法制备的乙烯共聚物。具体实施方式茂金属络合物用于制备乙烯共聚物的单中心(singlesite)茂金属络合物,特别是本发明中限定的由式(i)所定义的络合物是不对称的,这意味着形成茂金属络合物的两个配体是不同的。本发明可以用式(i)的茂金属络合物来实现其中m是hf或与zr的混合物,条件是式i的络合物按摩尔计超过50%具有m=hfx是σ配体r彼此相同或不同并且可以是饱和的直链或支链c1-c10烷基、c5-c10芳基、c6-c20烷基芳基或c6-c20芳基烷基基团,其可以任选地含有至多2个杂原子或硅原子r1是c6-c10芳基或c6-c20烷基芳基基团,任选地含有至多2个杂原子或硅原子或c4-c10杂芳基基团r2是式(ii)的c4-c20环烷基基团,任选地在β-位带有烷基取代基其中r’可以彼此相同或不同,并且可以是氢或被定义为r且n为1-17在式(i)中,每个x可以相同或不同,为σ配体,优选为氢原子、卤素原子、r3基团、or3基团、oso2cf3基团、ocor3基团、sr3基团、nr32基团或pr32基团,其中r3是直链的或支链的,环状的或非环状的,c1-c20-烷基、c2-c20-烯基、c2-c20-炔基、c6-c20-芳基、c7-c20-烷基芳基或c7-c20-芳基烷基自由基(radical);任选地含有属于第14-16族的杂原子或是sir33、sihr32或sih2r3。r3优选为c1-6-烷基、苯基或苄基基团。术语卤素包括氟、氯、溴和碘基团,优选氯基团。更优选地,每个x独立地为卤素原子或r3基团或or3基团,其中r3为c1-6-烷基、苯基或苄基基团。最优选x是甲基、苄基或氯基团。优选两个x基团相同。m优选为hf。r彼此相同或不同,并且可以是饱和的直链或支链c1-c10烷基、c5-c10芳基、c6-c20烷基芳基或c6-c20芳基烷基基团,其可以任选地包含至多2个杂原子或硅原子。优选r彼此相同或不同,并且可以是饱和的直链或支链c1-c6烷基、c6-c10芳基、c6-c20烷基芳基或c6-c20芳基烷基基团,其不含杂原子或硅原子。更优选所有r是相同的并且为饱和的直链或支链c1-c6烷基或c6-c10芳基,甚至更优选所有r是相同的并且为饱和的直链或支链c1-c6烷基。最优选所有r是相同的并且均为c1烷基基团。r1是c6-c10芳基或c6-c20烷基芳基基团,任选地含有至多2个杂原子或至多2个硅原子或是c4-c10杂芳基基团。c6-c20烷基芳基基团是指被一个或多个烷基基团取代的c6-c10芳基基团,所述多个烷基基团可以相同或不同,其中烷基基团取代基中的c原子数在导致c6-c20烷基芳基基团的范围内。r1的非限制性实例为苯基、对甲苯基、对异丙基苯基,3,5-二甲基苯基,β-萘基,4-(n,n-二甲基氨基)苯基,4-吡啶基,β-噻吩基,4-甲基-噻吩基等。优选r1是c6-c10芳基或c6-c12烷基芳基基团,其不含杂原子或硅原子或c4-c8杂芳基基团。更优选r1是未取代的苯基(即c6芳基),或被一至两个c1-c6烷基基团取代的苯基(即c7-c18烷基芳基基团)。甚至更优选r1是苯基、对-甲苯基或对-异-丙基苯基。最优选r1是苯基或对异丙基苯基。r2是式(ii)的c4-c20环烷基基团,任选地在β-位带有烷基取代基其中r’可以彼此相同或不同,并且可以是氢或定义为r且n为1-17。优选r2是式(ii)的c4-c10环烷基基团,其中n为1-7,并且r’可以彼此相同或不同并且可以是氢或饱和的直链或支链c1-c6烷基或c6-c10芳基。更优选r2是式(ii)的c4-c10环烷基基团,其中n为1-7,并且r’可以彼此相同或不同且可以是氢或饱和的直链或支链c1-c4烷基基团。r2的非限制性实例是环丁基、3,3-二甲基环丁基、环戊基、环己基、3,3,5,5-四甲基环己基等。最优选r2是式(ii)的c4-c8环烷基基团,其中n为1-5并且所有r’都是氢。式(i)的络合物的非限制性实例是1.(苯基)(环己基)亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二氯化铪2.(苯基)(环己基)亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二甲基铪3.(苯基)(环己基)亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二苄基铪4.(苯基)(环戊基)亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二氯化铪5.(苯基)(环戊基)亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二甲基铪6.(苯基)(环戊基)亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二苄基铪7.(苯基)(环丁基)亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二氯化铪8.(苯基)(环丁基)亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二甲基铪9.(4-异丙基苯基)(环己基)亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二氯化铪10.(4-异丙基苯基)(环己基)亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二甲基铪11.(4-异丙基苯基)(环戊基)亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二氯化铪12.(4-异丙基苯基)(环戊基)亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二甲基铪13.(4-异丙基苯基)(环丁基)亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二氯化铪14.(4-异丙基苯基)(环丁基)亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二甲基铪15.(3,5-二-异丙基苯基)(环己基)亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二氯化铪16.(3,5-二-异丙基苯基)(环己基)亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二甲基铪17.(3,5-二-异丙基苯基)(环戊基)亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二氯化铪18.(3,5-二-异丙基苯基)(环戊基)亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二甲基铪19.(3,5-二-异丙基苯基)(环丁基)亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二氯化铪20.(3,5-二-异丙基苯基)(环丁基)亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二甲基铪进一步优选的实例是,上述络合物以二苄基作为x取代基,而不是以二甲基或二氯化物作为x取代基。甚至更优选的是二甲基和二苄基络合物。助催化剂为了形成活性催化种类,通常需要使用本领域熟知的助催化剂。本发明需要使用含硼的助催化剂。感兴趣的基于硼的助催化剂包括含有硼酸根3+离子的硼化合物,即硼酸盐化合物。这些化合物通常含有下式的阴离子:(z)4b-(iii)其中z是任选取代的苯基衍生物,所述取代基是卤代c1-6-烷基或卤素基团。优选的选择是氟或三氟甲基。最优选地,所述苯基基团是全氟化的。这种离子助催化剂优选含有非配位阴离子,例如四(五氟苯基)硼酸盐。合适的抗衡离子是质子化胺或苯胺衍生物或磷离子。它们可具有通式(iv)或(v):nq4+(iv)或pq4+(v)其中q独立地为h、c1-6-烷基、c3-8环烷基、苯基c1-6-亚烷基-或任选取代的苯基(ph)。任选的取代基可以是c1-6-烷基,卤素或硝基。可存在一个或超过一个这样的取代基。因此,优选的取代的ph基团包括对位取代的苯基,优选甲苯基或二甲基苯基。优选至少一个q基团是h,因此优选的化合物是下式的那些化合物:nhq3+(vi)或phq3+(vii)优选的苯基c1-6-烷基-基团包括苄基。因此合适的抗衡离子包括:甲基铵、苯铵、二甲基铵、二乙基铵、n-甲基苯胺、二苯基铵、n,n-二甲基苯铵、三甲基铵、三乙基铵、三-正-丁基铵、甲基二苯基铵、对-溴-n,n-二甲基苯铵或对-硝基-n,n-二甲基苯铵,尤其是二甲基铵或n,n-二甲基苯铵。使用吡啶鎓作为离子是进一步的选择。感兴趣的磷离子包括三苯基磷、三乙基磷、二苯基磷、三(甲基苯基)磷和三(二甲基苯基)磷。更优选的抗衡离子是三苯甲基(cph3+)或其类似物,其中ph基团被官能化以带有一个或多个烷基基团。因此,本发明中使用的高度优选的硼酸盐包括四(五氟苯基)硼酸根离子。根据本发明可使用的优选离子化合物包括:三丁基铵四(五氟苯基)硼酸盐,三丁基铵四(三氟甲基苯基)硼酸盐,三丁基铵四-(4-氟苯基)硼酸盐,n,n-二甲基环己基铵四-(五氟苯基)硼酸盐,n,n-二甲基苄基铵四(五氟苯基)硼酸盐,n,n-二甲基苯铵四(五氟苯基)硼酸盐,n,n-二(丙基)铵四(五氟苯基)硼酸盐,二(环己基)铵四(五氟苯基)硼酸盐,三苯基碳鎓四(五氟苯基)硼酸盐(triphenylcarbeniumtetrakis(pentafluorophenyl)borate),二茂铁四(五氟苯基)硼酸盐。优选三苯基碳鎓四(五氟苯基)硼酸盐,n,n-二甲基环己基铵四(五氟苯基)硼酸盐、n,n-二甲基苄基铵四(五氟苯基)硼酸盐或n,n-二甲基苯铵四(五氟苯基)硼酸盐。更优选的硼酸盐是三苯基碳鎓四(五氟苯基)硼酸盐和n,n-二甲基苯铵四(五氟苯基)硼酸盐。最优选n,n-二甲基苯铵四(五氟苯基)硼酸盐。还可以加入烷基铝化合物。合适的烷基铝化合物是式(viii)air3的化合物,其中r是直链或支链的c2-c8-烷基基团。优选的烷基铝化合物是三乙基铝、三-异丁基铝、三-异己基铝、三-正辛基铝和三-异辛基铝。助催化剂的合适用量是技术人员公知的。硼与茂金属的金属离子的摩尔比可以在0.5:1至10:1mol/mol的范围内,优选1:1至10:1,特别是1:1至5:1mol/mol。甚至更优选的是硼与茂金属的金属离子的摩尔比为1:1至小于2:1mol/mol,例如1:1至1.8:1或1:1至1.5:1。催化剂制备本发明的茂金属络合物与一种或多种助催化剂组合使用,作为用于高温溶液聚合方法中乙烯和c4-10α-烯烃共聚单体聚合的催化剂体系。本发明的催化剂体系可以用作均相催化剂或非均相催化剂,优选用作均相催化剂体系。适用于本发明的均相或非负载催化剂体系可以在溶液中制备,例如在烃溶剂(如己烷、环己烷、庚烷、轻石脑油或甲苯)中,通过将所述茂金属(作为固体或作为溶液)与所述一种或多种助催化剂接触,例如硼烷或硼酸盐作为烃稀释剂中的浆料或预先溶解在芳族溶剂中,或优选通过将催化剂组分先后直接加入聚合反应器中来形成。聚合物使用本发明的催化剂体系制备的聚合物是乙烯和c4-10α-烯烃共聚单体的共聚物,所述c4-10α-烯烃共聚单体例如为1-丁烯,1-己烯,4-甲基-1-戊烯,1-辛烯等。优选使用丁烯,己烯或辛烯,更优选使用辛烯作为共聚单体。这种聚合物中的共聚单体含量可以达到45mol%,优选为1-40mol%,更优选为1.5-35mol%,甚至更优选为2-25mol%。聚合物的密度(根据iso1183-187测定)在0.850g/cm3-0.930g/cm3的范围内,优选在0.850g/cm3-0.920g/cm3的范围内,更优选在0.850g/cm3-0.910g/cm3的范围内。本发明聚合物的mw/mn值小于5,例如,在2.0-4.5的范围内。待制备的聚合物的熔点(根据iso11357-3:1999用dsc测定)低于130℃,优选低于120℃,更优选低于110℃,并且最优选低于100℃。聚合本发明的催化剂体系用于在高温溶液聚合方法中于高于100℃的温度下制备以上限定的乙烯共聚物。基于本发明,这种方法基本上是基于在液态烃溶剂中聚合单体和合适的共聚单体,其中所得的聚合物是可溶的。所述聚合在高于聚合物的熔点的温度下进行,其结果是获得聚合物溶液。闪蒸该溶液以将聚合物与未反应的单体和溶剂分离。然后回收该溶剂并在该方法中再循环。溶液聚合方法因其反应器停留时间短(与气相或淤浆方法相比)而为人所知,因此,允许非常快的品位转变和在短的生产周期内生产大范围产品的显著灵活性。根据本发明,使用的溶液聚合方法是一种高温溶液聚合方法,其使用高于100℃的聚合温度。优选所述聚合温度至少为110℃,更优选至少为150℃。所述聚合温度可高达250℃。根据本发明所用的溶液聚合方法中的压力优选在10-100巴的范围内,优选15-100巴,更优选20-100巴。所用的液态烃溶剂优选为c5-12-烃,其可以是未取代或被c1-4烷基基团取代,例如戊烷、甲基戊烷、己烷、庚烷、辛烷、环己烷、甲基环己烷和氢化石脑油。更优选使用未取代的c6-10-烃溶剂。优点新的催化剂体系,其包含组分(i)和(ii),可有利地用于在高温溶液聚合方法中的乙烯共聚。如果用于高温溶液聚合方法中的乙烯共聚,则根据本发明的催化剂体系显示出高生产率和高的溶解度、分子量能力和共聚单体引入能力。应用由本发明的催化剂体系制备的聚合物可用于各种最终制品,例如管材、薄膜(浇铸或吹塑薄膜)、纤维、模塑制品(例如注塑、吹塑、滚塑制品)、挤出涂料等。现在将参考以下非限制性实施例说明本发明实施例方法dsc温度调制的dsc实验在以调制模式操作的tainstrumentsq2000dsc中进行,并根据iso11357-1用铟、锡和锌校准。将约5mg样品置于铝盘中。最初将温度升至180℃,然后如标准dsc中那样以10℃/min降至-88℃。然后通过温度调制扫描升高温度,加热速率为2℃/min,每60秒调制0.32℃。从可逆热流热谱图中测量玻璃转变温度作为转变时的反转点。如果通过tg(dsc)来评估聚合物组成,则使用以下内部相关性:c8(wt%)=(tg(℃)+19.16)/-1.059zr和hf测定(icp法)催化剂的元素分析通过取在干冰上冷却的质量m的固体样品来进行。通过将样品溶解在硝酸(hno3,65%,5%v)和新鲜去离子(di)水(5%v)中,使样品稀释至已知体积v。然后将该溶液加入氢氟酸(hf,40%,3%v)中,用去离子水稀释至最终体积v,并使其稳定2小时。使用thermoelementalicap6300电感耦合等离子体-光学发射光谱仪(icp-oes)在室温下进行分析,其使用空白(5%hno3、3%hf的去离子水溶液)和6个标准品进行校准,所述6个标准品为含有0.5ppm、1ppm、10ppm、50ppm、100ppm和300ppm的al与0.5ppm、1ppm、5ppm、20ppm、50ppm和100ppm的hf和zr的5%hno3、3%hf的去离子水溶液。在分析之前,立即使用所述空白和100ppmal,50ppmhf、zr标准品进行“再斜率(resloped)”校准,运行质量控制样品(20ppmal、5ppmhf、zr的5%hno3、3%hf的去离子水溶液)以确认所述再斜率(reslope)。所述质量控制(qc)样品也在每第5个样品之后和在安排的分析组结束时运行。使用282.022nm线和339.980nm线监测铪的含量,并使用339.198nm线监测锆的含量。当在icp样品中al浓度在0-10ppm(仅校准至100ppm)时,通过167.079nm线监测铝的含量,而al浓度高于10ppm时,通过396.152nm线监测铝的含量。报告的值是取自相同样品的三个连续等分试样的平均值,并通过将样品的原始质量和稀释体积输入软件中来关联回到原始催化剂。通过nmr光谱定量共聚单体含量使用定量核磁共振(nmr)光谱定量聚合物的共聚单体含量。使用brukeradvanceiii500nmr光谱仪,对于1h和13c分别在500.13mhz和125.76mhz下操作,记录在熔融状态下的定量13c{1h}nmr光谱。使用13c优化的7mm魔角旋转(mas)探头在150℃下记录所有光谱,用氮气作为所有的气动。将约200mg物料填充到外径7mm的氧化锆mas转子中并以4khz旋转。选择此设置主要是为了快速鉴定和精确定量所需的高灵敏度。[1],[2],[3],[4]采用标准单脉冲激发,利用3s的短循环延迟的瞬态noe[5],[1]和rs-hept退耦方案。[6],[7]每个光谱获得总共1024(1k)个瞬态。选择该设置是因为其对低共聚单体含量的高灵敏度。对定量13c{1h}nmr光谱进行处理、积分,并使用通用光谱分析自动化程序确定定量特性。所有化学位移内部参考于30.00ppm处的本体(bulk)亚甲基信号(δ+)。[8]观察到对应于1-辛烯引入的特征信号[8],[9],[10],[11],[12]和基于聚合物中存在的所有其他单体计算出所有共聚单体含量。观察到由分离的1-辛烯引入所产生的特征信号,即eeoee共聚单体序列。使用38.32ppm处的信号积分来定量分离的1-辛烯引入。该积分分配至未能分辨的信号,分别对应于分离的(eeoee)和分离的双非连续(eeoeoee)1-辛烯序列的*b6位点及*βb6b6位点。为了补偿两个*βb6b6位点的影响,使用在24.7ppm处ββb6b6位点的积分:还观察到连续的1-辛烯引入产生的特征信号,即eeooee共聚单体序列。使用分配至ααb6b6位点占每个共聚单体的报告位点数在40.48ppm处的信号积分,来定量这种连续的1-辛烯引入:oo=2*iααb6b6还观察到由分离的非连续1-辛烯引入产生的特征信号,即eeoeoee共聚单体序列。使用分配至ββb6b6位点占每个共聚单体的报告位点数在24.7ppm处的信号积分,来定量这种分离的非连续1-辛烯引入:oeo=2*iββb6b6还观察到由分离的三连续1-辛烯引入产生的特征信号,即eeoooee共聚单体序列。使用分配至ααγb6b6b6位点占每个共聚单体的报告位点数在41.2ppm处的信号积分,来定量这种分离的三连续1-辛烯引入:ooo=3/2*iααγb6b6b6没有观察到指示其他共聚单体序列的其他信号,仅基于分离的(eeoee)、分离的双连续(eeooee)、分离的非连续(eeoeoee)和分离的三连续(eeoooee)1-辛烯共聚单体序列的量来计算总的1-辛烯共聚单体含量:o总=o+oo+oeo+ooo观察到由饱和端基产生的特征信号。使用22.84ppm和32.23ppm处的两个分辨信号的平均积分来定量这种饱和端基。将22.84ppm积分分配给分别对应于1-辛烯和饱和链端的2b6位点和2s位点的未能分辨的信号。32.23ppm积分分配给分别对应于1-辛烯和饱和链端的3b6位点和3s位点的未能分辨的信号。为了补偿2b6和3b61-辛烯位点的影响,使用了总1-辛烯含量:s=(1/2)*(i2s+2b6+i3s+3b6-2*o总)使用30.00ppm处的本体亚甲基(本体(bulk))信号的积分来定量乙烯共聚单体含量。该积分包括来自1-辛烯的γ和4b6位点以及δ+位点。基于本体积分计算总乙烯共聚单体含量,并补偿观察到的1-辛烯序列和端基:e总=(1/2)*[i本体+2*o+1*oo+3*oeo+0*ooo+3*s]应该注意的是,对于存在分离的三重引入(eeoooee)1-辛烯序列的本体积分无需补偿,因为欠量和过量的乙烯单元的数量相等。然后如下计算聚合物中1-辛烯的总摩尔分数:fo=(o总/(e总+o总)以标准方式由摩尔分数计算1-辛烯的总共聚单体引入重量百分比:o[wt%]=100*(fo*112.21)/((fo*112.21)+((1-fo)*28.05))[1]klimke,k.,parkinson,m.,piel,c.,kaminsky,w.,spiess,h.w.,wilhelm,m.,macromol.chem.phys.2006;207:382.[2]parkinson,m.,klimke,k.,spiess,h.w.,wilhelm,m.,macromol.chem.phys.2007;208:2128.[3]castignolles,p.,graf,r.,parkinson,m.,wilhelm,m.,gaborieau,m.,polymer50(2009)2373[4]nmrspectroscopyofpolymers:innovativestrategiesforcomplexmacromolecules,chapter24,401(2011)[5]pollard,m.,klimke,k.,graf,r.,spiess,h.w.,wilhelm,m.,sperber,o.,piel,c.,kaminsky,w.,macromolecules2004;37:813.[6]filip,x.,tripon,c.,filip,c.,j.mag.resn.2005,176,239[7]griffin,j.m.,tripon,c.,samoson,a.,filip,c.,andbrown,s.p.,mag.res.inchem.200745,s1,s198[8]j.randall,macromol.sci.,rev.macromol.chem.phys.1989,c29,201.[9]liu,w.,rinaldi,p.,mcintosh,l.,quirk,p.,macromolecules2001,34,4757[10]qiu,x.,redwine,d.,gobbi,g.,nuamthanom,a.,rinaldi,p.,macromolecules2007,40,6879[11]busico,v.,carbonniere,p.,cipullo,r.,pellecchia,r.,severn,j.,talarico,g.,macromol.rapidcommun.2007,28,1128[12]zhou,z.,kuemmerle,r.,qiu,x.,redwine,d.,cong,r.,taha,a.,baugh,d.winniford,b.,j.mag.reson.187(2007)225gpc:分子量平均值,分子量分布和多分散指数(mn,mw,mw/mn)聚合物的分子量平均值(mw,mn),分子量分布(mwd)及其宽度(该宽度由多分散指数描述,pdi=mw/mn(其中mn是数均分子量,mw是重均分子量))通过高温尺寸排阻色谱(ht-sec)在具有3个串联的plgelolexis(300×7.5mm,polymerlaboratories)柱的polymerlaboratoriesplxt-20rapidgpcpolymeranalysissystem(包括泵,折射率检测器和粘度检测器)上于160℃确定。使用含有丁基化羟基甲苯(0.5g/l)和irganox1010(20mg/l)的1,2,4-三氯苯作为洗脱剂,流速为1.0ml/min。基于聚乙烯标准品(polymerlaboratories,mp=5.310g/mol至mp=1.510.000g/mol)计算分子量。使用polymerlaboratoriesplxt-220自动样品处理系统作为自动进样器。样品的浓度为2-4mg聚合物/mltcb。相对共聚单体竞聚率r的确定液相中的乙烯浓度可以认为是恒定的,因为在聚合过程中通过进料乙烯使总压保持恒定。聚合结束时溶液中的c8/c2比通过从聚合物测得的组成中减去聚合物中引入的辛烯的量来计算(%wt1-辛烯)然后如下计算对于每种催化剂的竞聚率,r:r=[(c8/c2)聚合物]/[(c8/c2)液相中的平均值]其中(c8/c2)液相中的平均值计算为((c8/c2)最终+(c8/c2)进料)/2铪络合物在己烷中的溶解度溶解度的测试方法:所有实验均在隔膜瓶中于惰性气氛下进行。1.在手套箱内,在隔膜瓶中称量10.5mg络合物。2.将1g己烷加入到固体络合物中3.在室温下搅拌20h后,检查溶液4.如果没有观察到不溶物质,重复步骤1和步骤3直到达到饱和并观察到不溶物质。化学品n,n-二甲基苯铵四(五氟苯基)硼酸盐(ab)(cas118612-00-3)购自boulder。将作为共聚单体的1-辛烯(99%,sigmaaldrich)在分子筛上干燥并在使用前用氮气脱气。在分子筛上干燥庚烷和癸烷(99.9%,sigmaaldrich)并在使用前用氮气脱气。催化剂制备实施例a)络合物制备本发明络合物(ic):(苯基)(环己基)亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二甲基铪步骤1:6-苯基-6-环己基富烯向由6.11g(267.7mmol)钠金属和160ml无水乙醇得到的乙醇钠溶液中,在10min内滴加环戊二烯22.0g(332.8mmol),并将得到的红色溶液在室温下搅拌0.5h。然后,加入25.0g(132.8mmol)苯基环己基酮,并将得到的深红色混合物在室温下搅拌22h。然后,将其倒入1000ml水中。用2mhcl酸化至ph约6.5后,用500ml二氯甲烷萃取产物。分离有机层,经na2so4干燥并减压浓缩。通过快速色谱法在硅胶60(40-63um,800ml;洗脱剂:己烷)上分离产物。该方法得到22.09g(70%)6-苯基-6-环己基富烯,其为浅红色油,在-30℃保存时完全固化。c18h20理论计算值:c,91.47;h,8.53。实测值:c,91.79;h,8.72。1hnmr(cdcl3):δ7.35-7.29(m,3h),7.19-7.14(m,2h),6.71(dt,j=5.4hz,j=1.6hz,1h),6.54(dt,j=5.4hz,j=1.6hz,1h),6.36(dt,j=5.3hz,j=1.5hz,1h),5.74(dt,j=5.3hz,j=1.7hz,1h),3.10(tt,j=11.8hz,j=3.2hz,1h),1.85-1.73(m,4h),1.66(dm,j=13.2hz,1h),1.37(qt,j=12.9hz,3.2hz,2h),1.30(qd,j=12.4hz,3.2hz,2h),1.06(qt,j=12.9hz,3.7hz,1h).步骤2:苯基-环己基-环戊二烯基-(2,7-二-叔丁基芴-9-基)甲烷1.nbuli,thf向冷却至-50℃的26.0g(93.38mmol)2,7-二-叔丁基芴的250mlthf溶液中,一次性加入38.4ml(93.31mmol)的2.43mnbuli的己烷溶液。该混合物在室温下搅拌过夜。向所得的橙色溶液中,于室温下一次性加入22.09g(93.46mmol)6-苯基-6-环己基富烯的200mlthf溶液。在室温下搅拌过夜后,将深红色反应混合物在冰浴中冷却,然后用8.8ml12mhcl的100ml水溶液淬灭。将所得的橙色混合物用1000ml水稀释,然后用500ml乙醚萃取。分离有机层,并经na2so4干燥。真空除去溶剂,得到橙色油,将其溶解于250ml正己烷中,并将所得溶液在真空下缓慢蒸发至体积约50ml。将得到的悬浮液用200ml正己烷稀释,并过滤(g3),得到33.17g(69%)1-苯基-1-环己基-1-环戊二烯基-1-(2,7-二-叔丁基芴基)甲烷,为白色粉末。c39h46理论计算值:c,90.99;h,9.01。实测值:c,91.34;h,9.38。1hnmr(cdcl3):δ8.18-6.16(多重峰组,总和12h),6.02-4.86(单峰组,总和3h),3.57-0.13(单峰和多重峰组,总和31h)。步骤3:(苯基)(环己基)亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二氯化铪向冷却至-78℃的10.17g(19.76mmol)1-苯基-1-环己基-1-环戊二烯基-1-(2,7-二-叔丁基芴-9-基)甲烷的250ml乙醚溶液中,一次性加入15.8ml(39.5mmol)2.5mnbuli的己烷溶液。该混合物在室温下搅拌过夜。将得到的含有橙色沉淀的浅橙色溶液冷却至-50℃,并加入6.33g(19.76mmol)hfcl4。将形成的混合物在室温下搅拌24h,然后蒸发至干。将残余物与100ml温热的甲苯一起搅拌,将形成的悬浮液通过玻璃料(g4)过滤,并将滤液蒸发至约35ml。在室温下沉淀过夜,过滤(g4)掉白色固体并弃去。将母液蒸发至约20ml,向残余物中加入20ml正己烷。过滤(g3)掉在-30℃过夜沉淀出的黄色固体,并真空干燥,得到3.20g(苯基)(环己基)亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二氯化铪。将母液蒸发至干,并将残余物溶于25ml正己烷。收集在-30℃由该溶液过夜沉淀出的固体并真空干燥。该方法得到另外的0.60g目标络合物。因此,在该合成中分离的(苯基)(环己基)亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二氯化铪的总产量为3.80g(25%)。c39h44cl2hf理论计算值:c,61.46;h,5.82。实测值:c,61.37;h,6.10。1hnmr(cdcl3):δ8.02(d,j=8.8hz,1h),7.95(d,j=8.8hz,1h),7.76(d,j=7.8hz,1h),7.68(s,1h),7.63(dd,j=8.8hz,j=1.2hz,1h),7.57-7.51(m,2h),7.48(dd,j=8.8hz,j=1.6hz,1h),7.45-7.41(m,1h),7.39(ddd,j=7.5hz,j=7.5hz,j=1.0hz,1h),6.36-6.31(m,1h),6.21-6.16(m,1h),5.96(s,1h),5.76-5.71(m,1h),5.55-5.50(m,1h),3.26(t,j=11.5hz,1h),2.33(d,j=12.9hz,1h),2.14(d,j=12.3hz,1h),1.94-1.83(m,2h),1.16(d,j=13.2hz,1h),1.66-1.56(m,1h),1.53-1.40(m,1h),1.41(s,9h),1.40-1.30(m,1h),1.12-1.01(m,1h),0.96(s,9h),0.91-0.79(m,1h).步骤4:(苯基)(环己基)亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二甲基铪向3.80g(4.99mmol)(苯基)(环己基)亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二氯化铪的50ml甲苯和50ml乙醚的混合物的溶液中,加入7.0ml(14.77mmol)2.11mmemgbr的乙醚溶液。将得到的混合物回流30min,然后蒸发至约25ml。将得到的混合物加热至80-90℃,并趁热通过玻璃料(g4)过滤以除去不溶的镁盐。另外用5×20ml温热的正己烷洗涤滤饼。将合并的滤液蒸发至几乎干燥,然后向残余物中加入20ml正己烷。将得到的混合物再次通过玻璃料(g4)过滤。母液蒸发至干,并将残余物溶解在7ml正己烷中。收集在-30℃由该溶液过夜沉淀的黄色粉末,并真空干燥。该方法得到3.05g(88%)纯的(苯基)(环己基)亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二甲基铪。c41h50hf理论计算值:c,68.27;h,6.99。实测值:c,68.49;h,7.22。1hnmr(cdcl3):δ8.08(d,j=8.6hz,1h),7.98(d,j=8.9hz,1h),7.73(d,j=7.6hz,1h),7.58-7.48(m,3h),7.45(td,j=7.6hz,j=1.5hz,1h),7.38-7.28(m,3h),6.28-6.21(m,1h),6.12-6.06(m,1h),5.86(s,1h),5.62-5.55(m,1h),5.33-5.27(m,1h),3.06(t,j=11.5hz,1h),2.28(d,j=12.8hz,1h),2.10(d,j=12.3hz,1h),1.90-1.75(m,2h),1.70(d,j=13.3hz,1h),1.65-1.20(m,1h),1.48-1.33(m,1h),1.40(s,9h),1.31-1.16(m,1h),1.08-0.92(m,1h),0.95(s,9h),0.77-0.62(m,1h),-1.79(s,3h),-1.93(s,3h).13c{1h}nmr(cdcl3,):δ150.30,147.62,140.00,132.05,129.17,127.31,126.95,126.75,124.03,123.70,122.93,122.79,120.91,120.54,119.29,117.90,116.35,115.93,112.41,111.17,109.62,101.41,99.72,76.78,57.85,43.58,38.06,37.13,35.47,34.72,31.56,30.72,29.02,28.27,27.36,26.91,26.70.比较络合物1(cc1)(苯基)(1-己基)亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二甲基铪步骤1:6-苯基-6-己基富烯向在冰浴中冷却的23.8g(125.1mmol)1-苯基庚烷-1-酮的50mlthf溶液中,滴加25.4g(150mmol,1.2当量)环戊二烯基溴化镁的150mlthf溶液。所得混合物回流3h,然后在室温下搅拌过夜,然后在冰浴中冷却,之后用10%hcl淬灭至ph~5-6。该混合物用3×150ml己烷萃取;合并的有机萃取物经na2so4干燥,然后蒸发至干,得到深红色油。采用快速色谱法在硅胶60(40-63μm;洗脱剂:己烷-乙酸乙酯=100:1,体积)上分离产物。该方法得到14.25g(48%)6-苯基-6-己基富烯,为微红色油。c18h22理论计算值:c,90.70;h,9.30。实测值:c,90.87;h,9.27。1hnmr(cdcl3):δ7.37-7.30(m,5h),6.63(ddd,j=5.3hz,j=2.0hz,j=1.5hz,1h),6.55(ddd,j=5.3hz,j=2.0hz,j=1.5hz,1h),6.46(ddd,j=5.3hz,j=2.0hz,j=1.5hz,1h),6.10(ddd,j=5.3hz,j=2.0hz,j=1.5hz,1h),2.90(t,j=7.6hz,2h),1.47-1.36(m,2h),1.34-1.15(m,6h),0.83(t,j=6.9hz,3h).13c{1h}nmr(cdcl3):δ154.81,143.31,140.87,131.71,131.32,129.42,127.97,127.73,123.86,120.90,36.25,31.51,29.54,29.13,22.46,13.97.步骤2:(苯基)(1-己基)亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二氯化铪向冷却至-50℃的14.88g(53.44mmol)2,7-二-叔丁基芴的200mlthf溶液中,一次性加入21.4ml(53.5mmol)2.5mnbuli的己烷溶液。该混合物在室温下搅拌过夜。将得到的微红色溶液冷却至-50℃,并一次性加入14.25g(59.78mmol)6-苯基-6-己基富烯的200mlthf溶液。于室温下搅拌过夜后,将深红色反应混合物在冰浴中冷却,然后用5ml12mhcl的200ml水溶液淬灭。将得到的黄色混合物用400ml二氯甲烷萃取。分离有机层,并将水层用150ml二氯甲烷萃取。将合并的有机萃取物经na2so4干燥,并通过硅胶60(40-63μm)垫过滤,该硅胶垫另外用2×50ml二氯甲烷洗涤。将合并的滤液蒸发至干,得到29.1g苯基-(1-己基)-环戊二烯基-(2,7-二-叔丁基芴-9-基)甲烷,其无需进一步纯化即可使用。向冷却至-78℃的16.2g(31.35mmol)苯基-(1-己基)-环戊二烯基-(2,7-二-叔丁基芴基)甲烷(如上制备)的250ml乙醚溶液中,一次性加入25.0ml(62.5mmol)2.5mnbuli的己烷溶液。将形成的混合物在室温下搅拌过夜,然后冷却至-50℃,并加入10.04g(31.35mmol)hfcl4。将得到的混合物在室温下搅拌24h,然后蒸发至干。残余物与200ml温热的甲苯一同搅拌,并将形成的悬浮液通过玻璃料(g4)过滤。将滤液蒸发至约40ml。分离在-30℃下从该混合物在一周内沉淀出的黄色结晶物质,用3×15ml的甲苯和己烷(1:3,体积)的混合物洗涤并真空干燥。该方法得到14.2g(59%)(苯基)(1-己基)亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二氯化铪。c39h46cl2hf理论计算值:c,61.30;h,6.07。实测值:c,61.53;h,6.26。1hnmr(cdcl3):δ8.01(d,j=8.9hz,1h),7.95(d,j=8.9hz,1h),7.82(dm,j=7.9hz,1h),7.70(s,1h),7.66-7.58(m,2h),7.58-7.52(m,1h),7.52-7.46(m,1h),7.44-7.36(m,2h),6.36-6.30(m,1h),6.23-6.17(m,1h),6.13(s,1h),5.80-5.74(m,1h),5.55-5.49(m,1h),3.13-2.97(m,1h),2.80-2.65(m,1h),1.64-1.44(m,3h),1.40(s,9h),1.36-1.18(m,5h),0.99(s,9h),0.85(t,j=7.9hz,3h).13c{1h}nmr(cdcl3,):δ152.11,149.53,143.32,130.53,128.46,128.26,127.17,127.05,124.17,124.14,123.97,123.49,122.63,120.25,119.84,119.65,118.84,118.79,117.87,116.58,114.58,99.64(两个共振),77.83,53.68,41.28,35.42,34.84,31.88,31.11,30.51,29.75,24.03,22.63,14.05.步骤3:(苯基)(1-己基)亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二甲基铪向3.82g(5.0mmol)(苯基)(1-己基)亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二氯化铪的50ml甲苯和50ml乙醚的混合物溶液中,加入7.0ml(14.77mmol)2.11mmemgbr的乙醚溶液。将得到的混合物回流30min,然后蒸发至约25ml。将得到的溶液加热至80-90℃,并趁热通过玻璃料(g3)过滤以除去不溶的镁盐。另外用2×20ml甲苯洗涤滤饼。合并的滤液蒸发至干,并向残余物中加入20ml己烷。将得到的悬浮液通过玻璃料(g4)再次过滤。将滤液蒸发至干,并将残余物溶解在7ml己烷中。收集在-40℃下由该溶液过夜沉淀出的黄色粉末,并真空干燥。该方法得到2.55g(71%)纯的(苯基)(1-己基)亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二甲基铪。c41h52hf理论计算值:c,68.08;h,7.25。实测值:c,68.11;h,7.46。1hnmr(cdcl3):δ8.07(d,j=8.9hz,1h),7.98(d,j=8.9hz,1h),7.81(dm,j=7.9hz,1h),7.63-7.56(m,2h),7.53-7.43(m,2h),7.38-7.30(m,3h),6.28-6.21(m,1h),6.14-6.08(m,1h),6.03(s,1h),5.65-5.59(m,1h),5.34-5.28(m,1h),2.92-2.81(m,1h),2.59-2.48(m,1h),1.52-1.40(m,3h),1.38(s,9h),1.34-1.17(m,5h),0.97(s,9h),0.84(t,j=7.0hz,3h),-1.76(s,3h),-1.90(s,3h).13c{1h}nmr(cdcl3,):δ150.18,147.73,144.31,130.72,128.04,127.90,127.20,126.59,124.49,123.74,122.90,122.73,121.01,120.61,119.11,117.56,116.32,115.83,112.85,111.08,109.58,100.94,100.57,76.51,53.50,41.19,38.16,37.39,35.32,34.75,31.93,31.35,30.75,29.86,24.12,22.68,14.08.比较络合物2(cc2):(苯基)(异丙基)亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二甲基铪步骤1:6-异丙基-苯基富烯向冰浴中冷却的14.82g(100mmol)异丙基苯基酮的40mlthf溶液中,滴加20.3g(120mmol,1.2当量)环戊二烯基溴化镁的120mlthf溶液。将得到的混合物回流3h,然后室温下搅拌过夜,最后,在冰浴中冷却并用10%hcl淬灭至ph~5-6。该混合物用3×150ml己烷萃取,并将合并的有机萃取物经na2so4干燥。在真空下除去溶剂,得到深红色油。通过快速色谱法在硅胶60(40-63μm;洗脱剂:己烷)上分离产物。该方法得到8.32g(42%)6-异丙基-6-苯基富烯,为浅橙色油,其在-30℃下储存期间完全结晶。c15h16理论计算值:c,91.78;h,8.22。实测值:c,92.00;h,8.41。1hnmr(cdcl3):δ7.36-7.30(m,3h),7.21-7.15(m,2h),6.70(ddd,j=5.3hz,j=1.9hz,j=1.5hz,1h),6.54(ddd,j=5.3hz,j=1.9hz,j=1.5hz,1h),6.37(ddd,j=5.3hz,j=1.9hz,j=1.5hz,1h),5.77(ddd,j=5.3hz,j=1.9hz,j=1.5hz,1h),3.50(sept,j=7.0hz,1h),1.14(d,j=7.0hz,6h).13c{1h}nmr(cdcl3):δ159.82,143.03,138.92,132.14,130.72,129.20,127.19,126.98,124.36,120.22,33.72,22.13.步骤2:(苯基)(异丙基)亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二氯化铪向冷却至-50℃的11.77g(42.27mmol)2,7-二-叔丁基芴的250ml乙醚溶液中,一次性加入17.4ml(42.28mmol)2.43mnbuli的己烷溶液。将该混合物在室温下搅拌过夜。将得到的橙色溶液冷却至-50℃,并一次性加入8.30g(42.29mmol)6-异丙基-6-苯基富烯的150ml乙醚溶液。在室温下搅拌过夜后,将深红色反应混合物冷却至-50℃,并将17.4ml(42.28mmol)2.43mnbuli的己烷溶液一次性加入。将该混合物在室温下搅拌过夜。将得到的深红色溶液冷却至-50℃,并加入13.55g(42.3mmol)hfcl4。将形成的混合物在室温下搅拌24h,然后蒸发至干,并将残余物用150ml温热的甲苯处理。将该混合物趁热通过玻璃料(g4)过滤,将滤液蒸发至约50ml,并将80ml正己烷加入残余物中。过滤沉淀的固体并弃去。将母液蒸发至几乎干燥,并将残余物用70ml正己烷研磨。过滤橙黄色的沉淀,得到5.80g(约19%)(苯基)(异丙基)亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二氯化铪,纯度约90%。通过从甲苯中重结晶粗产物得到分析纯的样品(2.51g,8%)。c36h40cl2hf理论计算值:c,59.88;h,5.58。实测值:c,60.07;h,5.73。1hnmr(cdcl3):δ8.01(d,j=8.8hz,1h),7.95(d,j=8.9hz,1h),7.78(dm,j=7.8hz,1h),7.69(s,1h),7.62(d,j=8.9hz,1h),7.59-7.51(m,2h),7.49(dd,j=8.8hz,j=1.2hz,1h),7.47-7.36(m,2h),6.37-6.30(m,1h),6.22-6.16(m,1h),5.99(br.s,1h),5.75-5.69(m,1h),5.57-5.51(m,1h),3.69(sept,j=6.7hz,1h),1.39(s,9h),1.31(d,j=6.7hz,3h),1.05(d,j=6.7hz,3h),0.97(s,9h).13c{1h}nmr(cdcl3,):δ152.22,149.49,137.95,131.85,129.31,127.77,127.42,124.11,124.06,123.96,123.43,122.28,120.46,119.93,119.81,118.99,118.56,118.52,116.70,115.03,100.10,99.06,78.64,58.17,35.40,34.84,32.74,31.11,30.51,18.34,17.89.步骤3:(苯基)(异丙基)亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二甲基铪向3.3g(约4.6mmol)粗(苯基)(异丙基)亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二氯化铪(从甲苯中重结晶后的残余物)的30ml甲苯和15ml乙醚的混合物溶液中,加入6.5ml(17.55mmol)2.7mmemgbr的乙醚溶液。将得到的混合物在室温下搅拌过夜。蒸馏出大部分乙醚,并将所得的混合物通过玻璃料(g4)过滤以除去不溶的镁盐。将滤液蒸发至约15ml,并再次通过玻璃料(g4)过滤。将母液蒸发至约5ml,加入15ml正己烷,并将得到的轻微混浊的溶液通过玻璃料(g4)过滤。收集在-30℃由该滤液过夜沉淀的黄色粉末,并真空干燥。该方法得到1.46g(约47%)(苯基)(异丙基)亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二甲基铪。c38h46hf理论计算值:c,66.99;h,6.81。实测值:c,67.14;h,7.02。1hnmr(cdcl3):δ8.09(d,j=8.8hz,1h),7.99(d,j=8.8hz,1h),7.75(dm,j=7.9hz,1h),7.57(br.s,1h),7.56-7.44(m,3h),7.39-7.29(m,3h),6.27-6.23(m,1h),6.13-6.08(m,1h),5.88(br.d,j=1.0hz,1h),5.59-5.55(m,1h),5.33-5.29(m,1h),3.55-3.43(m,1h),1.38(s,9h),1.22(d,j=6.7hz,3h),0.97-0.93(s和d,总和12h),-1.79(s,3h),-1.92(s,3h).络合物元素分析(icp法):络合物hf(%)纯度zr(%)杂质cc199.610.39ic99.510.49比较实施例c0二苯基亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二甲基铪根据文献(hopf,a,kaminsky,w.,催化通讯(catalysiscommunications)2002;3:459)合成二苯基亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二氯化铪。向3.78g(5.0mmol)二苯基亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二氯化铪的50ml甲苯和50ml乙醚的混合物溶液中,加入7.0ml(14.77mmol)2.11mmemgbr的乙醚溶液。将所得的混合物回流30min,然后蒸发至约25ml。将得到的混合物加热至80-90℃,并趁热通过玻璃料(g4)过滤以除去不溶的镁盐。另外用5×20ml温热的己烷洗涤滤饼。将合并的滤液蒸发至约5ml,然后向残余物中加入20ml己烷。收集从该溶液沉淀出的黄色粉末并真空干燥。该方法得到3.14g(88%)纯的二苯基亚甲基(环戊二烯基)(2,7-二-叔丁基芴-9-基)二甲基铪。c41h44hf理论计算值:c,68.85;h,6.20。实测值:c,69.10;h,6.37。1hnmr(cdcl3):δ8.07(d,j=8.9hz,2h),7.95(br.d,j=7.9hz,2h),7.85(br.d,j=7.9hz,2h),7.44(dd,j=8.9hz,j=1.5hz,2h),7.37(td,j=7.6hz,j=1.2hz,2h),7.28(td,j=7.6hz,j=1.2hz,2h),7.24-7.17(m,2h),6.26(s,2h),6.20(t,j=2.7hz,2h),5.45(t,j=2.7hz,2h),1.03(s,18h),-1.90(s,6h).13c{1h}nmr(cdcl3,):δ148.46,145.75,129.69,128.63,128.46,126.73,126.54,123.29,122.62,120.97,118.79,116.09,111.68,107.76,101.56,76.47,57.91,37.61,34.88,30.84.b)聚合方法聚合试验已经在装有底阀的125ml反应器中于160℃和30巴(g)的总压力下进行。已经测试了液相中不同c8/c2重量比和不同催化剂装填,以找到在10分钟聚合期间保证几乎恒定的温度和压力的最佳量。·活化方法n,n-二甲基苯铵四(五氟苯基)硼酸盐已用于所有实验。tea(在isopare中7x10-2mol/kg)已被用作所有实验的清除剂,并且其用量已基于特定的实验设计(doe)与b/hf比率一起优化。通过将络合物和活化剂分别溶解在4ml甲苯中进行活化。然后将硼酸盐溶液注入反应器进料管线中,随后注入络合物溶液,并在通过氮气超压注入反应器之前使二者接触几秒钟。·聚合方法向反应器中填充所需量的溶剂(isopare)、清除剂(优选量0.035mmoltea)和1-辛烯。选择溶剂和单体的量以在聚合条件下具有80ml的初始液体体积。然后将反应器加热,并用乙烯(25-28巴(g))小心加压。当条件稳定时,将乙烯压力调节至30巴(g),并将混合物在750rpm下搅拌10分钟,以确定剩余的乙烯吸收。此后,催化剂(用4ml甲苯溶解)和助催化剂(也用4ml甲苯溶解)在注入管线中合并,并立即通过氮气超压注入反应器中。然后通过进料乙烯使压力保持恒定,并在10分钟后通过加入终止剂(etoh或co2)淬灭聚合。然后将反应器放空,降低温度并打开容器。将溶液排入含有几毫克(~500ppm基于所制备的共聚物)的irganox1076的铝盘中。然后将该盘置于通风良好的通风橱下,直至挥发物蒸发。根据聚合物表征段落中记载的方法,通过ht-sec和dsc分析所收集的残余物质。表1:乙烯/1-辛烯溶液共聚生产率结果发明实施例ie1-ie3和比较实施例ce1-ce6表1显示,基于生产率结果,当用硼酸盐ab作为助催化剂活化时,与己基(cc1)或异丙基(cc2)作为桥中的取代基相比,环己基作为在桥(ic)中的取代基引起更高的生产率(错误!未找到参考来源。)。对于催化剂装填0.07mg和停留时间10min,催化剂体系ic/ab显示出具有111kg-po/g-催化剂的最高生产率。对于相同的c8含量(或相同的平均c8/c2),在三种络合物之间没有观察到分子量能力和反应性的显著明显差异(图2-4)。在实验(doe)方法设计之后,通过开展一组实验来进一步研究ic/ab催化剂体系的性能。使用两个因素tea量和b/hf比(参见表2)。在这些实验中,已经评估了对生产率和聚合物性质(mw和c8引入)的影响。聚合温度为160℃,总压力为30巴(g)。结果报告在表2中,35μmoltea和1.5b/hf摩尔比获得了最佳生产率。表2:ic/ab体系ie2和ie4至ie12的乙烯/1-辛烯溶液共聚的聚合结果络合物在脂肪烃中的溶解度也是一个重要的性质,因为络合物优选作为溶在这种溶剂中的溶液进料到反应器中。该进料模式确保了反应器中催化剂浓度的更高精确度和更好的活化效率。本发明络合物在己烷中的溶解度示于表3中。表3铪络合物在己烷中的溶解度如表3所示,与络合物c0相比,络合物ic具有更高的溶解度。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1