增强功能性髓鞘产生的方法和组合物与流程
本申请要求2016年12月8日提交的美国临时专利申请第62/431,787号和2017年8月8日提交的美国临时专利申请第62/542,660号的申请日权益,上述每个申请的全部内容通过引用并入本发明。
发明背景
髓鞘相关病症影响数以百万计的人,对受影响个人及其家庭造成沉重的发病和死亡负担。脑白质营养不良是遗传性髓鞘相关病症,在美国总体上每7,500名新生儿中有一名受其影响。这些病症缺少疾病改善疗法,并且不可避免地在儿童期和青春期导致严重的发病和死亡。若干种常见脑白质营养不良具有已知的基因突变,导致中枢神经系统(cns)中的少突胶质细胞对神经元轴突的髓鞘形成(髓鞘包裹)不良。
佩梅病(pelizaeusmerzbacherdisease,pmd)是特别严重的脑白质营养不良,其导致四个月大时的认知和运动缺陷以及儿童期或成年早期的死亡。在更极端的情况中,患者在出生后两周内出现症状,从来不能学会走路或讲话,并且在10岁之前死于该疾病。
不幸的是,对许多这些病症的病理过程仍然知之甚少,并且几乎没有疾病改善疗法。因此,迫切需要对影响中枢神经系统髓鞘的病症的治疗方法。
技术实现要素:
本发明所述的实施方案涉及使用基因疗法或基因组工程治疗髓鞘相关病症的组合物和方法。
因此在一个方面中,本发明提供了一种产生使功能性髓鞘的产生增强的细胞的方法,所述方法包括如下对细胞进行基因修饰:(i)修饰内源性plp1基因以降低其抑制髓鞘产生的能力;(ii)修饰内源性plp1基因调控元件以降低其促进plp1表达的能力;(iii)修饰内源性plp1基因调控元件以提高其抑制plp1表达的能力;或(iv)修饰内源性plp1基因产物或促进plp1表达的plp1调控元件的基因产物以降低plp1表达水平,其中所述细胞产生功能性髓鞘或者是生成或分化为产生功能性髓鞘的细胞的祖细胞。
在一些实施方案中,产生的细胞通过降低细胞中plp1相关毒性来增强髓鞘产生。在一些实施方案中,修饰内源性plp1基因或plp1基因调控元件减轻了细胞中plp1相关的细胞应激。
在一些实施方案中,修饰内源性plp1基因可包括引入如下突变:其降低内源性plp1基因的表达或导致plp1转录物的降解(例如,通过无义介导的衰变)。
在一些实施方案中,内源性plp1基因或内源性plp1基因调控元件包含点突变,并且其中所述修饰内源性plp1基因或内源性plp1基因调控元件包括将点突变纠正为野生型序列。
在一些实施方案中,内源性plp1基因调控元件是plp1增强子或启动子。
在一些实施方案中,内源性plp1基因调控元件的基因修饰可包引入小的插入或缺失(插入缺失,indel)以改变plp1基因调控元件的活性,或引入较大的plp1的外显子缺失。
在一些实施方案中,基因修饰可包括靠近外显子1中起始密码子处或在plp1中前三个外显子中任何地方的大的缺失。在示例性实施方案中,基因修饰可包括在plp1的外显子3的5’末端的大的缺失。
在其他实施方案中,修饰内源性plp1基因调控元件可包括引入插入缺失或较大缺失以改变plp1基因调控元件(诸如plp1增强子或启动子)的活性。在一些方面,修饰内源性plp1基因调控元件以降低其促进plp1转录的能力。例如,修饰可包括plp1增强子或启动子的中断。
在一些实施方案中,内源性plp1基因是引起有害疾病的突变plp1基因。
在一些实施方案中,修饰内源性plp1基因或plp1基因调控元件可使用核酸酶进行。核酸酶可包括锌指核酸酶(zfn)、tale-效应子(talen)、crispr/cas系统或ngago系统。
在一些实施方案中,核酸酶可包括第2类crispr/cas系统。例如,第2类crispr/cas系统可包括ii型基于cas9的crispr系统或基于v型cpf1的crispr系统。
在一些实施方案中,在外显子1或外显子3处用crispr/cas系统核酸酶修饰plp1基因。在一些实施方案中,在外显子3的5’末端用crispr/cas系统核酸酶修饰plp1基因。在一些实施方案中,通过中断外显子1中的起始密码子而用crispr/cas系统核酸酶修饰plp1基因。
在一些实施方案中,修饰plp1基因产物或plp1调控元件的基因产物包括向细胞递送基因沉默剂。在一些实施方案中,基因沉默剂可包括rnai构建物(诸如,sirna、shrna或mirna或者可被转录以生产这些的构建物)。
在一些实施方案中,基因沉默剂可包括反义寡核苷酸(aso)。
在一些实施方案中,进行基因修饰的细胞显示增强的髓鞘产生(例如,由于细胞中plp1相关的毒性降低)。
在一些实施方案中,所述方法包括将细胞与包含核酸酶或基因沉默剂的递送载体相接触。
在一些实施方案中,递送载体是aav载体、腺病毒载体或慢病毒载体。
在一些实施方案中,所述方法包括:(a)将细胞与第一aav载体和第二aav载体以及任选地第三aav载体相接触,所述第一aav载体包含编码功能性ii型crispr-cas9的核酸(诸如cas9或cas9直系同源cdna),第二aav载体包含对内源性plp1基因或内源性plp1基因调控元件中的靶位点有特异性的向导rna(sgrna)序列,且第三aav载体包含用于纠正或替换内源性plp1基因或内源性plp1基因调控元件的缺陷或突变部分的供体核酸序列;或者(b)将细胞与第一aav载体以及任选地第三aav载体相接触,所述第一aav载体包含编码功能性ii型crispr-cas9的核酸(诸如cas9或cas9直系同源cdna)和向导rna(sgrna)序列,所述向导rna(sgrna)序列以顺式编码并且对内源性plp1基因或内源性plp1基因调控元件中的靶位点有特异性,且所述第三aav载体包含用于纠正或替换内源性plp1基因或内源性plp1基因调控元件的缺陷或突变部分的供体核酸序列。
在一些实施方案中,第一aav载体进一步任选地以5’→3’的取向包含以下元件中的一种或多种:i)5’aav末端反向重复(itr);ii)启动子和任选的增强子;iii)编码功能性ii型crispr-cas9的cas9cdna;iv)多腺苷酸化信号;和v)3’aav末端反向重复(itr)。
在一些实施方案中,启动子和任选的增强子可以是泛素型或组成型启动子和任选的泛素型或组成型增强子;可调控、可诱导或可脱抑制的启动子和任选的可调控、可诱导或可脱抑制的增强子;组织特异性启动子和任选的组织特异性增强子;病毒启动子和任选的病毒增强子;在受精卵、opc、nsc或少突胶质细胞中有活性的启动子;病毒启动子、任选地cmv启动子或病毒增强子;哺乳动物β肌动蛋白启动子;禽类β肌动蛋白启动子;哺乳动物u6启动子;或人u6启动子。
在一些实施方案中,第二或第三aav载体(当存在时)进一步包含以下元件中的一种或多种,任选地以5’→3’的取向:i)5’aavitr;ii)启动子和任选的增强子;iii)向导rna序列;iv)填充物或填充核酸序列;和v)3’aavitr。
在一些实施方案中,第三aav载体(当存在时)进一步包含以下元件中的一种或多种,任选地以5’→3’的取向:i)5’aavitr;ii)5’剪接受体(sliceacceptor)位点;iii)供体核酸序列;iv)多腺苷酸化信号;和v)aav3’itr。
在一些实施方案中,第一、第二和/或第三aav载体包含选自以下任何血清型的vp1、vp2或vp3衣壳:aav1、aav2、aav3、aav4、aav5、aav6、aav7、aav8、aav9、aav10、aav11或者其混合物、变体或衍生物。
在一些实施方案中,5’aavitr选自以下中任一种:aav1、aav2、aav3、aav4、aav5、aav6、aav7、aav8、aav9、aav10、aav11、或者其嵌合体或融合体、或者其中3’aavitr选自以下中任一种:aav1、aav2、aav3、aav4、aav5、aav6、aav7、aav8、aav9、aav10、aav11或者其嵌合体或融合体。
在一些实施方案中,在体外、体内或离体接触细胞。
本发明的另一方面提供了一种组合物,其包含如本发明所述任一实施方案的第一和第二aav载体(以及任选地第三aav载体)。
本发明的另一方面提供了一种药物组合物,其包含如本发明所述的组合物。
本发明的另一方面提供了一种基因修饰细胞。所述细胞可如下进行基因修饰:(i)修饰内源性plp1基因以降低其抑制髓鞘产生的能力;(ii)修饰内源性plp1基因调控元件以降低其促进plp1表达的能力;(iii)修饰内源性plp1基因调控元件以提高其抑制plp1表达的能力;或(iv)修饰内源性plp1基因产物或促进plp1表达的plp1调控元件的基因产物以降低plp1表达水平,其中细胞产生功能性髓鞘或者是产生或分化为产生功能性髓鞘的细胞的祖细胞。
在一些实施方案中,细胞选自神经干细胞(nsc)、少突胶质祖细胞(opc)、神经元细胞和神经胶质细胞,诸如少突胶质细胞、星形胶质细胞、室管膜细胞或小神经胶质细胞,优选nsc、opc和少突胶质细胞,更优选nsc或opc。
本发明的另一方面涉及一种基因修饰细胞,其来自或分化自如下进行基因修饰的细胞:(i)修饰内源性plp1基因以降低其抑制髓鞘产生的能力;(ii)修饰内源性plp1基因调控元件以降低其促进plp1表达的能力;(iii)修饰内源性plp1基因调控元件以提高其抑制plp1表达的能力;或(iv)修饰内源性plp1基因产物或促进plp1表达的plp1调控元件的基因产物以降低plp1表达水平,其中细胞产生功能性髓鞘或者是生成或分化为产生功能性髓鞘的细胞的祖细胞。
本发明的另一方面涉及包含如上所述的基因修饰细胞的组合物以及治疗受试者的髓鞘相关病症的方法。所述方法包括对受试者施用如下进行基因修饰的细胞:(i)修饰内源性plp1基因以降低其抑制髓鞘产生的能力;(ii)修饰内源性plp1基因调控元件以降低其促进plp1表达的能力;(iii)修饰内源性plp1基因调控元件以提高其抑制plp1表达的能力;或(iv)修饰内源性plp1基因产物或促进plp1表达的plp1调控元件的基因产物以降低plp1表达水平,其中细胞在受试者中产生功能性髓鞘或者是产生或分化为产生功能性髓鞘的细胞的祖细胞。
在相关方面,本发明提供了一种治疗受试者的髓鞘相关病症的方法,所述方法包括根据本发明的方法对受试者的细胞进行基因修饰,从而在受试者中产生功能性髓鞘,其中髓鞘相关病症优选特征在于异常的plp1基因活性和/或表达。
在一些实施方案中,髓鞘相关病症选自多发性硬化(ms)、视神经脊髓炎(nmo)、横贯性脊髓炎、慢性炎性脱髓鞘性多发性神经病、格林-巴利综合征、进行性多灶性脑白质病(pml)、脑脊髓炎(epl)、脑桥中央髓鞘溶解症(cpm)、肾上腺脑白质营养不良、亚历山大病、佩梅病(pmd)、沃勒变性、视神经炎、肌萎缩性脊髓侧索硬化症(als)、亨廷顿病、阿尔茨海默病、帕金森病、脊髓受伤、外伤性脑损伤、辐射后损伤、化疗的神经并发症、中风、急性缺血性视神经病、维生素e缺乏症、孤立性维生素e缺乏症综合征、巴-科综合征、马毕二氏综合征、三叉神经痛、marie-charcot-tooth病、贝尔面瘫和脑白质营养不良。
在其他实施方案中,髓鞘相关病症可以是脑白质营养不良,诸如选自下组之一:具有髓鞘碱性蛋白缺陷的18q综合征、急性播散性脑脊髓炎(adem)、急性播散性脑白质炎、急性出血性脑白质病、肾上腺脑白质营养不良(ald)、肾上腺脊髓神经病(amn)、成年发病的常染色体显性遗传脑白质营养不良(adld)、成人葡聚糖体病、艾卡迪综合征(aicardi-goutieressyndrome)、亚历山大病、常染色体显性弥漫性脑白质病伴轴索球样变(hdls)、常染色体显性晚发性脑白质病、卡纳万病、儿童共济失伴弥漫性中枢神经系统脱髓鞘(cach或消融性脑白质病)、常染色体显性脑动脉病伴随皮质下梗死和脑白质病(cadasil)、脑视网膜微血管造影伴钙化和囊肿、脑腱黄瘤病(ctx)、儿童共济失调伴中枢神经系统脱髓鞘(cach)、颅骨干骺端发育不良伴脑白质病、囊性脑白质病(rnaet2相关)、超长链脂肪酸-4的延长(elongationofverylong-chainfattyacids-4,elovl4;pseudo-sjogren-larsson)、无临床症状的广泛性脑白质异常、表现为小脑性共济失调和痴呆的家族性成年发作的脑白质营养不良、家族性脑白质营养不良伴成年发作的痴呆和糖脂存储异常、脂肪酸2-羟化酶缺陷、岩藻糖苷贮积症、福山型先天性肌营养不良症、半乳糖唾液酸贮积症、球状细胞脑白质营养不良(克拉伯病)、gm1神经节苷脂贮积症、gm2神经节苷脂贮积症(戴萨克斯症)、遗传性成年发作脑白质营养不良刺激的慢性进行性多发性硬化、遗传性弥漫性脑白质病伴轴索球样变(hdls)、脱髓鞘伴基底神经节和小脑萎缩(h-abc)、脱髓鞘、促性腺激素分泌不足、生殖官能不良和牙发育不全(4h综合征)、脂膜样骨发育不良伴脑白质营养不良(nasu病)、异染性脑白质病变(mld)、巨脑性脑白质营养不良伴皮质下囊肿(mlc)、神经轴脑白质病伴轴索球样变(遗传性弥漫性脑白质病伴球样变–hdls)、新生儿肾上腺脑白质营养不良(nald)、眼指齿发育不良伴脑白质异常、正色性脑白质营养不良伴色素性神经胶质、卵巢性脑白质营养不良综合征、佩梅病(x-连锁的痉挛性截瘫)、雷弗素姆病、sjogren-larsson综合征、嗜苏丹性脑白质营养不良、vanderknaap综合征(空泡性脑白质营养不良伴皮质下囊肿或mlc)、消融性白质病(vwm)或儿童共济失调伴弥散性中枢神经系统脱髓鞘(cach)、x-连锁的肾上腺脑白质营养不良(x-ald)、齐薇格谱系(zellwegerspectrum):齐薇格、新生儿肾上腺脑白质营养不良和婴儿雷弗素姆病。
在一些实施方案中,髓鞘相关病症包括佩梅病(pmd)。
在一些实施方案中,所述方法将受试者的寿命恢复到未患有髓鞘相关病症的对照受试者的至少30%、40%、50%、60%、70%、80%、90%或约100%。
在一些实施方案中,所述方法减轻了受试者的与所述髓鞘相关病症有关的至少一种症状。
在一些实施方案中,所述方法将受试者的功能恢复到未患有髓鞘相关病症的对照受试者的至少30%、40%、50%、60%、70%、80%、90%或约100%,优选地,所述功能为运动协调、运动或轴突传导速度。
在一些实施方案中,细胞选自下组:基因修饰的nsc、opc、神经元细胞、少突胶质细胞、星形胶质细胞、室管膜细胞和小神经胶质细胞,优选nsc、opc或少突胶质细胞。
在一些实施方案中,内源性plp1基因或其基因调控元件或其部分(诸如不超过4.8、4.5、4.0、3.5、3.0、2.5、2.0、1.5或1.0kb的部分)被失活、中断、纠正或替代。
在一些实施方案中,受试者是哺乳动物,诸如人(例如,年龄小于20岁、15岁、10岁、5岁、3岁、2岁、1岁、6个月、3个月、1个月、2周、1周、3日或1日的人)。
本发明所公开的所有特征可以任何组合相结合。除非另外明确说明,说明书中公开的每个特征均可由用于相同、等同或类似目的替代性特征来代替。
附图说明
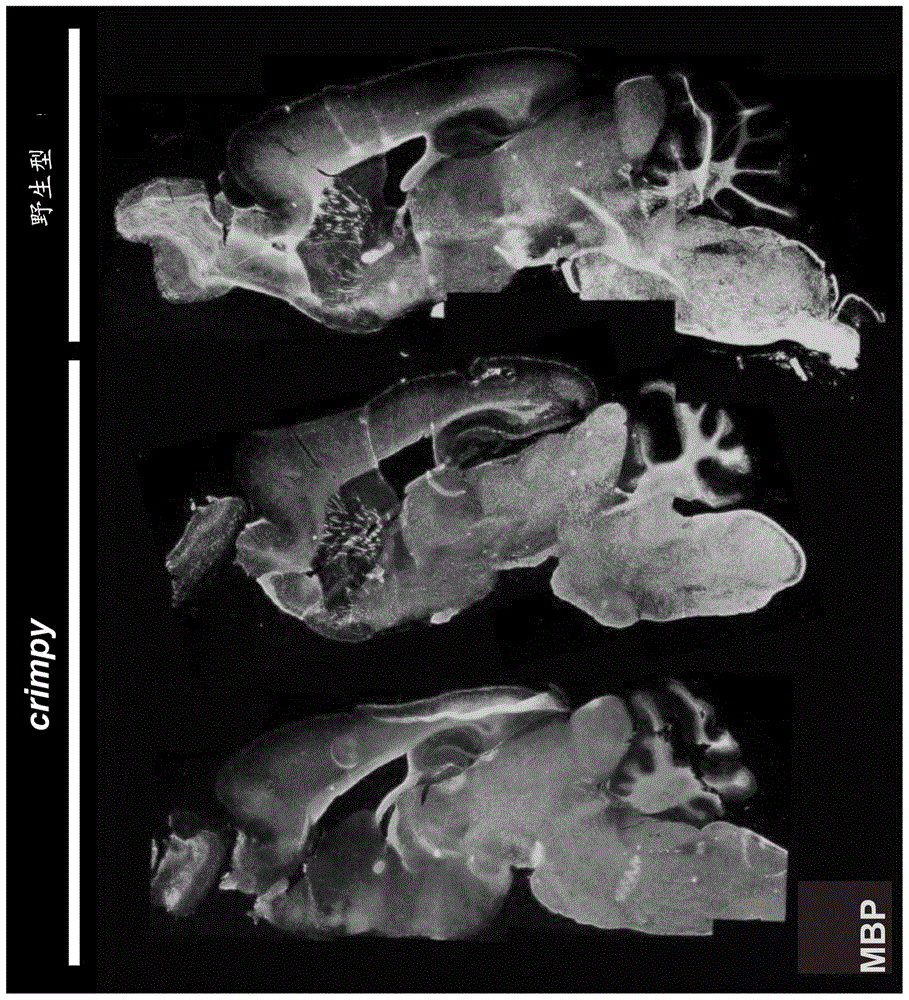
图1是显示了plp1-插入缺失纠正的-jimpy(或crimpy)pmd模型小鼠中的中枢神经系统的完全髓鞘形成的组织学分析图,其在mbp(髓鞘碱性蛋白)染色中与野生型无法区分。
图2是显示小鼠plp1基因中jimpy基因突变的位置以及最终导致少突胶质细胞死亡的所得基因产物的示意图(不按比例)。该图的底部显示jimpy小鼠的寿命严重缩短,中位存活期约为23日。
图3显示了jimpy小鼠脑中严重的髓鞘形成减少。注意,与野生型对照物相比,在出生后第19日(p19),jimpy小鼠脑切片中标记成熟少突胶质细胞的髓鞘碱性蛋白(mbp)染色显著减少。jimpy小鼠还显示了同一年龄的意向性震颤和共济失调的神经症状(数据未显示)。
图4和5是示意图,其显示了通过靶向外显子3的crisprspcas9/双向导rna(sgrna)敲除jimpy小鼠受精卵中plp1基因的示例性方法,以产生crispr敲除的jimpy(cr-impy[或crimpy])后代小鼠。图4显示了靶向外显子3中位点的sgrna的相对位置。图5显示了产生用于接收sgrna和spcas9mrna的jimpy雄性受精卵的一般试验方法。成功的由crispr/cas9介导的jimpyplp1基因的敲除导致由代孕宿主雌性娩出的plp1为空的雄性cr-impy初代(founder)的出生。与亲本品系杂交的两代产生了子代小鼠以进一步表征。在出生后21日(p21),当大部分jimpy小鼠显示严重的神经症状或死亡时,cr-impy小鼠缺乏明显的表型(数据未示出)。
图6显示了cr-impy小鼠与jimpy对照物和野生型对照物相比具有恢复的寿命。
图7显示了通过检测mbp的全脑ihc检测,cr-impy小鼠显示了成熟少突胶质细胞的恢复。比较了出生后19日和出生后6个月。
图8显示了用于评估cr-impy小鼠的运动协调的旋转杆(rotarod)测试的示意图,其中通过在旋转杆加速时测量从旋转杆下落的时间来定量运动协调。测量在出生后19日(p19)、出生后2个月和出生后6个月,在野生型、jimpy和cr-impy小鼠中在每个时间点进行。不同值之间的统计学显著性由p值表示。结果显示与野生型和jimpy小鼠相比,cr-impy小鼠的运动协调有所恢复。
图9显示了用于评估cr-impy小鼠自主活动的旷场测试的示意图,其中通过自动视频跟踪5分钟来测量在盒子中行经的总距离来定量自主活动。测量在出生后19日(p19),出生后2个月和出生后6个月,在野生型、jimpy和cr-impy小鼠中在每个时间点进行。不同值之间的统计学显著性通过p值表示。结果显示与野生型和jimpy小鼠相比,cr-impy小鼠中的自主活动有所恢复。
图10显示了视神经传导速度测试以及较快和较慢的传导峰(分别为第一峰和第二峰)的代表性结果。与无髓鞘和较小直径的轴突相比,有髓鞘和较大直径的轴突通常具有较快的传导。在出生后19日(p19),分别通过第一峰和第二峰测量的快和慢传导速度在野生型、jimpy小鼠和cr-impy小鼠中任何两种之间均在统计学上显著不同。然而,在出生后6个月,尽管所有jimpy小鼠已死亡,在野生型和cr-impy小鼠之间没有观察到区别。
图11显示了在出生后6个月时,在野生型和cr-impy小鼠之间,在视神经em图像中没有可辨识的区别。
图12是显示本发明一个实施方案的示意图,其中crispr-cas9介导的基因沉默被传递给出生后的大脑,例如,通过编码sacas9和单一sgrna的aav病毒载体,其可至少部分纠正患者脑中的突变opc。如此纠正的opc将产生少突胶质细胞,其将及时胜过任何突变少突胶质细胞,因此经过一段时间将提供充分恢复的髓鞘形成和患者神经元功能。
发明详述
定义
为方便起见,在这里汇总了在说明书、实施例和所附权利要求中使用的一些术语。除非另外定义,本发明所用的所有技术和科学术语都具有与本申请所述领域中普通技术人员所通常理解的相同含义。
冠词"一/一个/一种(a和an)"在本发明中用于指代一个或多于一个(即,指代至少一个)的该冠词的语法受体。举例来说,“一个(a)元件”表示一个元件或者多于一个元件。
术语“包括/包含/具有/含有"(“comprise”,“comprising”,“include”,“including”,“have”和“having”)以包含性的开放含义使用,意味着可包含其他要素。术语"诸如"、"例如"在本发明中是非限制性的,并且仅用于说明目的。“包括”和“包括但不限于”可互换使用。
除非上下文明确表示相反,本发明所用的术语“或”应理解为表示“和/或”。
本发明所用的术语“约”或“大致”是指量、水平、值、数字、频率、百分比、尺寸、大小、数量、重量或长度相对于参考量、水平、值、数字、频率、百分比、尺寸、大小、数量、重量或长度的变化至多为15%、10%、9%、8%、7%、6%、5%、4%、3%、2%或1%。在一个实施方案中,术语“约”或“大致”是指相对参考的量、水平、值、数字、频率、百分比、尺寸、大小、数量、重量或长度±15%、±10%、±9%、±8%、±7%、±6%、±5%、±4%、±3%、±2%或±1%的量、水平、值、数字、频率、百分比、尺寸、大小、数量、重量或长度的范围。
短语“肠胃外施用”和“经肠胃外给药”是本领域认可的术语,并且包括除了肠内和局部施用之外的施用方式,诸如注射,包括但不限于,静脉内、肌肉内、胸膜内、血管内、心包内、动脉内、鞘内、囊内、眶内、心内、皮内、腹膜内、经气管内、皮下、表皮下、关节内、囊下、蛛网膜下、脊柱内和胸骨内(intrastemal)注射和输注。
术语“治疗”是本领域公知的,并且包括抑制受试者的疾病、障碍或病症,例如,阻止其进展;以及缓解疾病、障碍或病症,例如,导致疾病、障碍和/或病症的逆转。治疗疾病或病症包括减轻特定疾病或病症的至少一个症状,即使潜在的病理学没有受到影响。
术语“预防”是本领域公知的,并且包括阻止可能易患疾病、障碍或病症但尚未被诊断患病的受试者的疾病、障碍或病症发作。预防与疾病相关的病症包括在疾病已被诊断出来但尚未诊断出病症之后阻止该病症。
术语“药物组合物”是指其形式适合用于施用给受试者的包含所公开的化合物的制剂。在优选实施方案中,药物组合物为散装或为单位剂型。单位剂型为包括以下的多种形式中的任何种类:例如,胶囊剂、iv包、片剂、气溶胶吸入器上的单泵或小瓶。单位剂量组合物中的活性成分量(例如,所公开的化合物或其盐的制剂)为有效量,并且根据所涉及的具体治疗而变。本领域技术人员将意识到,有时必须根据患者的年龄和病症对剂量做常规改动。剂量也将取决于施用途径。设想了多种途径,包括口服、肺部、直肠、肠胃外、透皮、皮下、静脉、肌肉内、腹膜内、鼻内、吸入等。用于局部或透皮施用的本发明所述化合物的剂型包括粉末剂、喷雾剂、软膏剂、糊剂、乳膏剂、洗剂、凝胶剂、溶液剂、贴剂、雾化化合物和吸入剂。在优选实施方案中,将活性化合物在无菌条件下与药学可接受的载体和与任何需要的防腐剂、缓冲剂或喷射剂混合。
短语“药学可接受的”是本领域公知的。在一些实施方案中,该术语包括如下的组合物、聚合物和其他材料和/或剂型:在合理的医学判断范围内,其适合用于与人和动物的组织接触而没有过多的毒性、刺激、过敏反应或者其他问题或并发症,与合理的利益/风险比相称。
短语“药学可接受的载体”是本领域公知的,并且包括,例如,药学可接受的材料、组合物或媒介物,诸如涉及将任何主题组合物从身体的一个器官或部分携带或输送到身体的另一个器官或部分的液体或固体的填料、稀释剂、赋形剂、溶剂或包封材料。每种载体都必须是“可相容的”,即与主题组合物的其他成分相容和对患者无害。在一些实施方案中,药学可接受的载体是无热原的。可用作药学可接受的载体的材料的一些实例包括:(1)糖,诸如乳糖、葡萄糖和蔗糖;(2)淀粉,诸如玉米淀粉和马铃薯淀粉;(3)纤维素及其衍生物,诸如羧甲基纤维素钠、乙基纤维素和醋酸纤维素;(4)粉末状黄蓍胶;(5)麦芽;(6)明胶;(7)滑石;(8)赋形剂,诸如可可油和栓剂蜡;(9)油,诸如花生油、棉籽油、葵花籽油、芝麻油、橄榄油、玉米油和大豆油;(10)二醇,诸如丙二醇;(11)多元醇,诸如甘油、山梨醇、甘露醇和聚乙二醇;(12)酯,诸如油酸乙酯和月桂酸乙酯;(13)琼脂;(14)缓冲剂,诸如氢氧化镁和氢氧化铝;(15)藻酸;(16)无热原水;(17)等渗盐水;(18)林格氏溶液;(19)乙醇;(20)磷酸盐缓冲溶液;和(21)其他用于药物制剂中的无毒可相容物质。
术语“预防性”或“治疗性”治疗是本领域公知的,并且包括向主体施用一种或多种主题组合物。如果是在临床表现出不想要的病症(例如,主体动物的疾病或其他不理想状态,诸如但不限于髓鞘形成障碍、髓鞘缺陷、髓鞘流失和髓鞘修复无效)之前施用,则治疗是预防性的,即,其保护主体免于发展出不想要的病症,但如果是在表现出不想要的病症之后,则治疗是治疗性的(即,其旨在减少、改善或稳定已有的不想要病症或其副作用)。
术语“治疗剂”、“药物”、“药品”和“生物活性物质”是本领域公知的,并且包括如下的分子和其他试剂:其是局部或全身性作用于患者或受试者以治疗疾病或病症的具有生物、生理或药理活性的物质。该术语包括但不限于其药学可接受的盐和前药。这样的试剂可以是酸性的、碱性的或者是盐;其可以是中性分子、极性分子或能够氢键结合的分子配合物;其可以是以下形式的前药:当施用给患者或受试者时能被生物激活的醚、酯、酰胺等。
短语“治疗有效量”或“药学有效量”是本领域公知的术语。在一些实施方案中,该术语是指以适用于任何药物治疗的合理利益/风险比产生一些所需效果的治疗剂的量。在一些实施方案中,该术语是指消除、减少或维持特定治疗方案的目标的必要或足够量。有效量可取决于诸如以下因素:所治疗的疾病或病症、所施用的特定靶向构建物、受试者的尺寸或者疾病或病症的严重性。本领域技术人员可经验性地确定特定化合物的有效量而不必进行过多实验。在一些实施方案中,用于体内使用的试剂(例如,本发明所述的组合物或基因修饰细胞)的治疗有效量取决于多种因素,包括:试剂从聚合物基质的释放速率,这将部分取决于聚合物的化学和物理特性;试剂的身份;施用方式和方法;以及聚合物基质中除该试剂之外加入的任何其他材料。
术语“核酸”、“核苷酸”、“多核苷酸”和“寡核苷酸”可互换使用,它们是指线性或环状构造以及单链或双链形式的脱氧核糖核苷酸或核糖核苷酸聚合物。对于本发明,这些术语不应解释为限制聚合物的长度。该术语可包含天然核苷酸以及在碱基、糖和/或磷酸盐部分(例如,硫代磷酸盐主链)进行修饰的核苷酸的已知类似物。通常,特定核苷酸的类似物具有相同的碱基配对特异性;即,a的类似物将与t碱基配对。
术语“多肽”、“肽”和“蛋白质”可互换地用于指代氨基酸残基的聚合物。该术语还用于其中一个或多个氨基酸为相应的天然氨基酸的化学类似物或修饰衍生物的氨基酸聚合物。
“功能域”是包括特定活性的多肽的结构域。功能域可具有的活性的非限制性实例为核酸酶活性、转录调控活性、病毒衣壳识别活性等。
“结合”是指大分子之间(例如,蛋白质和核酸之间)的序列特异性非共价相互作用。并非所有结合相互作用的组件都需要是序列特异性的(例如,与dna主链中的磷酸盐残基接触),只要相互作用整体上是序列特异性的即可。这样的相互作用通常可表征为解离常数(kd)为10-6m-1或更低。“亲和性”是指结合的强度:较高的结合亲和性与较低的kd相关。
“结合蛋白”是能结合另一个分子的蛋白质。结合蛋白可结合,例如,dna分子(dna-结合蛋白)、rna分子(rna-结合蛋白)和/或蛋白质分子(蛋白-结合蛋白)。在蛋白-结合蛋白的情况中,其可自身结合(形成均二聚体、均三聚体等)和/或其可结合不同蛋白或多种不同蛋白的一个或多个分子。结合蛋白可具有多于一种类型的结合结合活性。例如,锌指蛋白具有dna-结合、rna-结合和蛋白-结合活性。
“锌指dna结合蛋白”(或结合结构域)是以序列特异性方式通过一个或多个锌指结合dna的蛋白质或较大蛋白质内的结构域,所述锌指是结合结构域中其结构通过锌离子配位来稳定的氨基酸序列区域。术语锌指dna结合蛋白经常简称为锌指蛋白或zfp。
“taledna结合结构域”或“tale”是包含一个或多个tale重复序列结构域/单元的多肽。该重复序列结构域涉及tale与其同源靶dna序列的结合。单一“重复序列结构域”(也称为“重复”)通常是长度为33-35个的氨基酸,并且显示与天然tale蛋白中的其他tale重复序列的至少一些序列同一性。
锌指和tale结合结构域可被“工程化”以结合预定的核苷酸序列,例如通过天然锌指或tale蛋白的识别螺旋区的工程化(改变一个或多个氨基酸)。因此,工程化dna结合蛋白(锌指或tale)是非天然蛋白质。工程化dna-结合蛋白的方法的非限制性实例是设计和选择。所设计的dna结合蛋白是非天然产生的蛋白质,其设计/组成主要得自理性规范。设计的理性规范包括应用替换规则和用于处理存储了现有zfp和/或tale设计信息和结合数据的数据库中的信息的计算机算法。参见,例如,美国专利6,140,081;6,453,242;6,534,261和8,585,526;也参见wo98/53058;wo98/53059;wo98/53060;wo02/016536和wo03/016496。
“选定的”锌指蛋白或tale是不曾在自然中发现的蛋白质,其产生主要源自经验方法,诸如噬菌体展示、相互作用陷阱或杂交选择。参见,例如,美国专利5,789,538;5,925,523;6,007,988;6,013,453;6,200,759;8,586,526;wo95/19431;wo96/06166;wo98/53057;wo98/54311;wo00/27878;wo01/60970;wo01/88197;wo02/099084。
通常,“crispr”(成簇的规律间隔的短回文重复序列)也称为spidr(间隔子穿插直接重复),其是指通常对特定细菌种群有特异性的dna基因座家族。crispr基因座包含在大肠杆菌中识别的不同类别的穿插短序列重复(ssr)(ishino等(1987)j.bacteriol.,169:5429-5433;和nakata等,j.bacteriol.(1989)171:3553-3556)以及相关基因。类似的穿插ssr已在以下中被鉴定:地中海嗜盐菌(haloferaxmediterranei)、化脓性链球菌(streptococcuspyogenes)、项圈藻(anabaena)和结核分枝杆菌(mycobacteriumtuberculosis)(参见,groenen等(1993)mol.microbiol.,10:1057-1065;hoe等(1999)emerg.infect.dis.,5:254-263;masepohl等(1996)biochim.biophys.acta1307:26-30;和mojica等(1995)mol.microbiol.,17:85-93)。crispr基因座与其他ssr的区别通常在于重复的结构,其已被命名为规律的短间隔重复(srsr)(janssen等(2002)omicsj.integ.biol.,6:23-33;和mojica等(2000)mol.microbiol.,36:244-246)。通常,重复是成簇出现的短元件,这些短元件由具有基本恒定长度的独特插入序列规律地间隔开(mojica等(2000),出处同上)。尽管重复序列在菌株之间高度保守,但穿插的重复的数量和间隔子区域的序列通常逐株不同(vanembden等,j.bacteriol.(2002)182:2393-2401)。crispr基因座已在多于40种原核生物中被鉴定:包括但不限于:气火菌属(aeropyrum)、火棒菌属(pyrobaculum)、硫化叶菌属(sulfolobus)、古球状菌属(archaeoglobus)、嗜盐小盒菌属(halocarcula)、甲烷杆菌属(methanobacteriumn)、产甲烷球菌属(methanococcus)、甲烷八叠球菌属(methanosarcina)、甲烷火菌属(methanopyrus)、火球菌属(pyrococcus)、嗜酸菌属(picrophilus)、thernioplasnia、棒状杆菌属(corynebacterium)、分支杆菌属(mycobacterium)、链霉菌属(streptomyces)、产液菌属(aquifrx)、卟啉单胞菌属(porphvromonas)、绿菌属(chlorobium)、thermu、芽孢杆菌属(bacillus)、利斯特菌属(listeria)、葡萄球菌属(staphylococcus)、梭菌属(clostridium)、高温厌氧杆菌属(thermoanaerobacter)、支原菌属(mycoplasma)、梭形杆菌属(fusobacterium)、azarcus、色素杆菌属(chromobacterium)、奈瑟氏菌属(neisseria)、亚硝化单胞菌属(nitrosomonas)、脱硫弧菌属(desulfovibrio)、土杆菌属(geobacter)、myrococcus、弯曲杆菌属(campylobacter)、沃林氏菌属(wolinella)、不动细菌属(acinetobacter)、欧文氏菌属(erwinia)、埃希氏菌属(escherichia)、军团杆菌属(legionella)、甲基球菌属(methylococcus)、巴斯德氏菌属(pasteurella)、发光杆菌属(photobacterium)、沙门氏菌属(salmonella)、黄单胞杆菌属(xanthomonas)、耶尔森氏菌属(yersinia)、密螺旋体属(treponema)和栖热孢菌属(thermotoga)。
“crispr系统”总体上是指涉及cpispr相关(“cas”)基因活性的表达或指导该活性的转录体和其他元件,包括编码cas基因的序列、tracr(反式激活crispr)序列(例如,tracrrna或活性部分tracrrna)、tracr-mate序列(包括“直接重复”,以及在内源性crispr系统背景下的tracrrna-处理的部分直接重复)、向导序列(在内源性crispr系统的背景下也称为“间隔子”)或来自crispr基因座的其他序列和转录物。在一些实施方案中,crispr系统的一个或多个元件来源于第1类i型或iii型crispr系统。在一些实施方案中,crispr系统的一个或多个元件来源于第2类ii型或v型crispr系统。在一些实施方案中,crispr系统的一个或多个元件来源于包含内源性crispr系统的特定生物体,诸如化脓性链球菌(streptococcuspyogenes)。通常,crispr系统的特征在于促进crispr复合物在靶序列位点处的形成的元件(在内源性crispr系统的背景下,也称为前间区序列)。在形成crispr复合物的背景下,“靶序列”是指如下的序列:向导序列被设计为与该序列具有互补性,其中靶序列与向导序列之间的杂交促进了crispr复合物的形成。不需要完全互补,条件是存在足够的互补性以引起杂交和促进crispr复合物的形成。靶序列可包含任何多核苷酸,诸如dna或rna多核苷酸。在一些实施方案中,靶序列位于细胞的细胞核或细胞质中。可用于重组进入包含靶序列的靶基因座的序列或模板被称为“编辑模板”或“编辑多核苷酸”或“编辑序列”。在本发明的多个方面,外源模板多核苷酸可被称为编辑模板。在本发明的一个方面,重组是同源重组。
“ngago”是被认为涉及基因沉默的原核argonaute蛋白。ngago来源于古细菌格氏嗜盐碱杆菌(natronobacteriumgregoryi)(参见,例如,gao等(2016)naturebiotechnology34,768-773)。“ngago系统”是所需的所有组件,包括,例如,用于ngago酶切割的单链向导dna。
“重组”是指在两个多核苷酸之间交换遗传信息的过程,包括但不限于通过非同源末端连接(nhej)的供体捕获和同源重组。对于本发明,“同源重组(hr)”是指发生在例如通过同源定向修复机制修复细胞中的双链断裂过程中的这种交换的特定形式。这一过程需要核苷酸序列同源性,使用“供体”分子来模板修复“靶”分子(即经历双链断裂的分子),并且被不同地称为“非-交叉基因转换”或“短链基因转换”,因为其导致供体到靶标的遗传信息传递。不希望受任何特定理论限制,这种传递可涉及形成于断裂的靶和供体之间的异源双链dna的错配纠正,和/或其中供体用于重新合成将会成为靶的一部分的遗传信息的“合成依赖性链退火(annealing)”,和/或相关过程。这种专门的hr通常导致靶分子序列的改变,从而使供体多核苷酸序列的一部分或全部被加入到靶多核苷酸中。
在本发明的方法中,如本发明所述的一个或多个靶核酸酶在靶序列(例如,细胞染色质)中在预定位点处切割(例如,产生一个或多个单链缺口和/或一个或多个双链断裂[dsb])。dsb可通过同源介导修复(例如,hdr)或通过非同源介导修复机制(例如,nhej)导致缺失和/或插入。缺失可包括任何数量的碱基对。类似地,插入可包括任何数量的碱基对,包括,例如,整合“供体”多核苷酸,其任选与断裂区域的核苷酸序列具有同源性。
在一些实施方案中,本发明的方法使用任何合适的核酸酶(诸如crispr/cas系统核酸酶)在靶基因(诸如plp1基因或其调控元件)中产生dsb,并且所得的修复(诸如nhej)产生了破坏plp1基因或其调控元件的功能的小插入或缺失(例如,插入缺失),导致plp1功能的降低或消除。在该实施方案中,本发明的方法不需要供体序列。
在其他实施方案中,在方法中可包括供体序列以修复或替换缺陷的plp1基因或其调控元件。供体序列可被物理整合,或者可选地,供体多核苷酸被用作经由同源重组修复断裂的模板,导致将供体中的全部或部分核苷酸序列引入细胞染色质中。因此,细胞染色质中的第一序列可被改变,并且在一些实施方案中,其可被转化为供体多核苷酸中存在的序列。因此,术语“替代”或“替换”的使用可被理解为表示将一个核苷酸序列替换为另一个(即,在信息意义上的序列替换),并且不需要一个多核苷酸对另一个的物理或化学替换。
在本发明所述的任何方法中,额外的锌指蛋白、talen、crispr/cas或ngago系统对可用于细胞内额外靶位点的额外(例如,两个或更多个)双链切割(例如,在相同的靶基因如plp1内,但在不同的靶向位点处)。
本发明所述的任何方法可用于插入任何尺寸的供体和/或通过靶向整合破坏了所关注的基因的表达来部分或完全失活细胞中的一个或多个靶序列。还提供了具有部分或完全失活基因的细胞系。
在本发明所述的任何方法中,外源核苷酸序列(“供体序列”或“转基因”)可包含与所关注的区域中的基因组序列同源但不相同的序列,从而刺激同源重组而在所关注的区域插入不同序列。因此,在一些实施方案中,与所关注的区域中的序列同源的供体序列部分显示与所替换的基因组序列具有约80-99%(或其间的任何整数)的序列同一性。在其他实施方案中,供体和基因组序列之间的同源性大于99%,例如,如同在超过100个连续碱基对的供体和基因组序列之间有1个核苷酸不同。在一些情况中,供体序列的非同源部分可包含不存在于所关注的区域中的序列,这样可将新序列引入到所关注的区域中。在这些情况中,非-同源序列通常侧接50-1,000个碱基对(或其间的任何整数值)或大于1,000的任何数量的碱基对的序列,所述序列与所关注的区域的序列同源或相同。在其他实施方案中,供体序列与第一序列非-同源,并且通过非-同源重组机制插入到基因组中。
“基因修饰”是指对核酸所做的修饰,该修饰使得核酸的序列与修饰之前的核酸相比有所改变。细胞的基因修饰是指改变细胞内的细胞核酸,包括对细胞中的内源性和/或外源核酸(诸如基因组dna和转录的mrna)进行基因修饰。基因修饰可包括细胞内的内源性和/或外源核酸的切割、缺失和插入,外源dna的插入,基因纠正和/或基因突变。
“切割”是指核酸(例如,dna或rna,诸如mrna)分子的共价主链的断裂。可通过多种方法引发切割,包括但不限于磷酸二酯键的酶或化学水解。单链切割和双链切割都是可以的,双链切割可作为两个不同的单链切割事件发生。dna切割可导致产生平端或交错末端。在一些实施方案中,融合多肽被用于靶向双链dna切割。
“切割半结构域”是如下的多肽序列:其与第二多肽(相同或不同)结合形成具有切割活性(优选双链切割活性)的复合物。术语“第一和第二切割半结构域”、“+/-切割半结构域”和“左右切割半结构域”可互换地用于指代二聚的切割半结构域对。
“工程化切割半结构域”是已进行修饰以与另一个切割半结构域(例如,另一个工程化切割半结构域)形成专性异二聚体的切割半结构域。也参见美国专利公开号2005/0064474、20070218528、20080131962和20110201055,上述文献通过引用以其整体并入本发明。
术语“序列”是指任何长度的核苷酸序列,其可以是dna或rna;可以是线性、环状或分枝的;并且可以是单链或双链的。
术语“供体序列”是指插入到基因组中的核苷酸序列。供体序列可具有任何长度,例如长度为2-100,000,000个核苷酸(或者其间或其上的任何整数值),优选长度为约100-100,000个核苷酸(或其间的任何整数),更优选长度为约2000-20,000个核苷酸(或其间的任何值),甚至更优选地,约5-15kb(或其间的任何值)。
“染色质”是包含细胞基因组核蛋白结构。细胞染色质包括核酸(主要为dna)和蛋白质(包括组蛋白和非组蛋白染色体蛋白)。大部分真核生物的细胞染色质以核小体形式存在,其中核小体核心包括与包含组蛋白h2a、h2b、h3和h4中各两个的八聚体相连的约150个碱基对的dna;以及在核小体核心之间延伸的接头dna(根据生物体具有不同的长度)。组蛋白h1的分子通常与接头dna相连。对于本发明,术语“染色质”意图包含所有类型的细胞核蛋白,包括原核生物和真核生物。细胞染色质包括染色体和附加型染色质(例如,线粒体dna)。
“染色体”是包含细胞的全部或部分基因组的染色质复合物。细胞的基因组通常以其染色体组型表征,其是包含细胞基因组的所有染色体的集合。细胞的基因组可包含一个或多个染色体。
“附加体”是复制核酸、核蛋白复合体或包含并非细胞染色体组型的一部分的核酸的其他结构。附加体的实例包括质粒和一些病毒基因组/载体(诸如aav载体和所编码的序列)。
“可达区(accessibleregion)”是细胞染色质中的位点,其中核酸中存在的靶位点可被识别靶位点的外源分子结合。不希望受任何特定理论限制,相信可达区是未被包含(packaged)到核小体结构中的区域。可达区的独特结构通常可通过其对化学和酶探针(例如,核酸酶)的敏感性来检测。
“靶位点”或“靶序列”是限定了结合分子将要结合的核酸部分的核酸序列,条件是存在结合的充分条件。
“外源”分子是通常不存在于细胞中但可通过一种或多种遗传、生物化学或其他方法引入细胞中的分子。“通常存在于细胞中”是相对于细胞的特定发育阶段和环境条件来确定的。因此,例如,仅在肌肉的胚胎发育期间存在的分子相对于成年肌肉细胞而言是外源分子。类似地,因热休克产生的分子相对于非热休克细胞而言是外源分子。外源分子可包括,例如,功能故障的内源性分子的功能形式或功能正常的内源性分子的功能故障形式。
外源分子可为小分子等,诸如通过组合化学方法所生成的;或者为大分子,诸如诸如蛋白质、核酸、糖类、脂质、糖蛋白、脂蛋白、多糖、上述分子的任何修饰衍生物或包含一种或多种上述分子的复合物。核酸包括dna和rna,可为单链或双链的;可为线性、分枝或环状的;并且可具有任何长度。核酸包括能够形成双链的那些,以及形成三链的核酸。参见,例如,美国专利5,176,996和5,422,251。蛋白质包括但不限于dna-结合蛋白、转录因子、染色质重塑因子、甲基化dna结合蛋白、聚合酶、甲基化酶、去甲基化酶、乙酰基转移酶、去乙酰化酶、激酶、磷酸酶、整合酶、重组酶、连接酶、拓扑异构酶、促旋酶和解旋酶。
外源分子可为与内源性分子相同类型的分子,例如,外源蛋白质或核酸。例如,外源核酸可包括被引入到细胞中的感染病毒基因组、质粒或附加体病毒,或通常不存在于细胞中的染色体。将外源分子引入细胞中的方法是本领域技术人员已知的,包括但不限于脂质-介导的转移(即,脂质体,包括中性和阳离子脂质)、电穿孔、直接注入、细胞融合、粒子轰击、磷酸钙共沉淀、deae-葡聚糖-介导的转移和病毒载体-介导的转移。外源分子还可为与内源性分子相同类型的分子,但来源于与细胞来源不同的物种。例如,人核酸序列可被引入到最初来源于小鼠或仓鼠的细胞系中。将外源分子引入植物细胞的方法是是本领域技术人员已知的,包括但不限于原生质体转化、碳化硅(例如,whiskerstm)、土壤杆菌-介导的转化、脂质-介导的转移(即,脂质体,包括中性和阳离子脂质)、电穿孔、直接注入、细胞融合、粒子轰击(例如,使用“基因枪”)、磷酸钙共沉淀、deae-葡聚糖-介导的转移和病毒载体-介导的转移。
相反,“内源性”分子是在特定发育阶段在特定环境条件下通常存在于特定细胞中的分子。例如,内源性核酸可包括染色体,线粒体、叶绿体或其他细胞器的基因组,或者天然附加型核酸。其他内源性分子可包括蛋白质,例如,转录因子和酶。
本发明所用的术语“外源核酸产物”包括多核苷酸和多肽产物,例如,转录产物(多核苷酸,诸如rna)和翻译产物(多肽)。
“融合”分子是其中(优选共价)连接了两个或更多个亚单位分子的分子。亚单位分子可为相同化学类型的分子或可为不同化学类型的分子。第一种类型融合分子的实例包括但不限于融合蛋白(例如,zfp或taledna-结合结构域与一个或多个激活结构域之间的融合)和融合核酸(例如,编码上文所述融合蛋白的核酸)。第二种类型融合分子的实例包括但不限于形成三链的核酸与多肽之间的融合以及小沟结合物与核酸之间的融合。
在细胞中表达融合蛋白可通过将融合蛋白递送到细胞中或者通过将编码融合蛋白的多核苷酸递送到细胞中,其中转录该多核苷酸并翻译转录物以生成融合蛋白。在蛋白质于细胞中的表达中也可涉及反式剪接、多肽切割和多肽连接。将多核苷酸和多肽递送到细胞中的方法在本发明中其他地方阐述。
对于本发明,“基因”包括编码基因产物(见下文)的dna区域以及调控基因产物产生的所有dna区域(基因调控元件),无论这种基因调控元件序列是否邻近编码序列和/或转录序列。因此,基因包括但不必限制于启动子序列、终止子、翻译调控序列(诸如核糖体结合位点和内部核糖体进入位点)、增强子、沉默子、绝缘子、边界元件、复制起点、基质附着位点和基因座控制区。
“基因表达”是指将基因中所含的信息转化到基因产物中。基因产物可为基因的直接转录产物(例如,mrna、trna、rrna、反义rna、核糖酶、结构rna或任何其他类型的rna)或通过翻译mrna产生的蛋白质。基因产物还包括通过诸如封端、多腺苷酸化、甲基化以及编辑的方法修饰的rna;和通过例如甲基化、乙酰化、磷酸化、泛素化、adp-核糖基化、豆蔻酰化和糖基化修饰的蛋白质。
基因表达的“调控”是指基因活性的改变。表达调控可包括但不限于基因活化和基因阻遏,以及诸如蛋白质和mrna的基因表达产物的活性和/或稳定性(例如,在转录、翻译和/或翻译后水平上的表达调控)。基因组编辑(例如,切割、改变、失活、随机突变)可用于调控表达,rnai或反义寡核苷酸(aso)-介导的mrna切割和/或翻译阻断也是如此。基因失活是指与不包含如本发明所述的zfp、tale、ngago或crispr/cas系统的细胞相比,基因表达的任何降低。因此,基因失活可以是部分或完全的。
“所关注的区域”是细胞染色质的任何区域,诸如,例如,其中需要结合外源分子的基因或者基因内或相邻的非-编码序列。结合可用于靶向dna切割和/或靶向重组。所关注的区域可存在于例如染色体、附加体、细胞器基因组(例如,线粒体、叶绿体)或感染病毒基因组中。所关注的区域可在基因的编码区内,在转录的非编码区内(例如,前导序列、尾随序列或内含子),或在编码区上游或下游的非转录区内。所关注的区域的长度可以小到单个核苷酸对或者多达2,000个核苷酸对或任何整数值的核苷酸对。
“真核”细胞包括但不限于真菌细胞(诸如酵母)、植物细胞、动物细胞、哺乳动物细胞和人细胞(例如,少突胶质细胞),包括干细胞(多能(pluripotent)和多效(multipotent))。
术语“可操作的连接”和“可操作地连接”(或“可操作地相连”)在涉及两个或更多个组成部分(诸如序列元件)并置时可互换使用,其中所述组成部分被布置为使得两个组成部分都正常工作并且允许至少一个组成部分能够介导至少一个其他组成部分所具有的功能的可能性。举例来说,如果转录调控元件序列响应于一个或多个转录调控因子的存在或不存在来控制编码序列的转录水平,则该转录调控元件序列(诸如启动子)可操作地连接编码序列。转录调控元件序列通常以顺式可操作地连接编码序列,但不必与其直接相邻。例如,增强子是可操作地连接编码序列的转录调控元件序列,即使它们不是连续的。
对于融合多肽,术语“可操作地连接”可指以下事实:每个组成部分与其他组成部分连接实施相同功能,如同其没有这样连接时那样。例如,对于其中zfp、tale、ngago或casdna-结合结构域融合到活化结构域的融合多肽,如果融合多肽中的zfp、tale、ngago或casdna-结合结构域部分能够结合其靶位点和/或其结合位点,同时活化结构域能够上调基因表达,则zfp、tale、ngago或casdna-结合结构域与活化结构域是可操作地连接的。当其中zfp、tale、ngago或casdna-结合结构域融合到切割结构域时,如果融合多肽中zfp、tale、ngago或casdna-结合结构域部分能够结合其靶位点和/或其结合位点,同时切割结构域能够在靶位点附近切割dna,则zfp、tale、ngago或casdna-结合结构域和切割结构域是可操作地连接的。
蛋白质、多肽或核酸的“功能片段”是其序列不同于全长蛋白质、多肽或核酸,但仍保留与全长蛋白质、多肽或核酸相同功能的蛋白质、多肽或核酸。功能片段可具有与相应的天然分子相比更多、更少或相同数量的残基和/或可包含一个或多个氨基酸或核苷酸置换。测定核酸功能(例如,编码功能、与另一种核酸杂交的能力)的方法是本领域公知的。类似地,测定蛋白质功能的方法是公知的。例如,可通过例如滤膜结合、电泳迁移率或免疫沉淀实验来测定多肽的dna-结合功能。可通过凝胶电泳测定dna切割。参见ausubel等,见上文。可通过例如免疫共沉淀、双杂合测试或互补(基因和生化的)来测定蛋白质与另一种蛋白质相互作用的能力。参见,例如,fields等(1989)nature340:245-246;美国专利5,585,245和pctwo98/44350。
“载体”能够将基因序列转移到靶细胞。通常,“载体构建物”、“表达载体”、和“基因转移载体”表示能够指导所关注的基因的表达并且能将基因序列转移到靶细胞中的任何核酸构建物。因此,该术语包括克隆和表达介质以及整合载体。
术语“受试者”和“患者”可互换使用,其是指哺乳动物,诸如人类患者和非人灵长类动物,以及实验动物,诸如,兔、犬、猫、大鼠、小鼠和其他动物。因此,本发明所用的术语“受试者”或“患者”表示可对其施用本发明的核酸酶、供体和/或基因修饰细胞的任何哺乳动物患者或受试者。本发明的受试者包含那些患有髓鞘相关障碍者。
“干细胞特性”是指任何细胞以类似干细胞的方式发挥作用的相对能力,即,任何特定干细胞可具有的全能(toti-potentcy)、多能(pluri-potentcy)或寡能(oligo-potentcy)的程度以及扩展或不确定的自我更新。
“增强功能性髓鞘产生的细胞”表示显示更多的髓鞘产生量(与未经修饰的细胞相比)的细胞和/或显示细胞所产生的髓鞘的功能性能力改善的细胞。
术语“插入缺失(indel)”是指生物体或细胞的核苷酸序列中的碱基的插入、缺失及其组合。插入(也称插入突变)是将一个或多个核苷酸碱基对添加到核苷酸序列中,而缺失是指其中去除了一部分核苷酸序列的突变。可插入或缺失任何数量的核苷酸,从一个碱基到整块染色体。
“无义介导的衰变”是指消除包含过早翻译终止密码子(ptc)的mrna转录物的真核细胞中的翻译-偶联机制。在哺乳动物细胞中,nmd也与前体-mrna剪接相关,因为在许多情况中,仅当ptc位于内含子上游时才发生强烈的mrna减少。
综述
本发明所述的实施方案涉及产生经基因修饰以破坏或失活蛋白脂质蛋白1(plp1)基因的方法以及其用于治疗人髓鞘相关障碍的方法。本申请部分基于在神经胶质细胞中和在受精卵中有效定点诱导plp1-失活的插入缺失的证明。已显示,核酸酶-介导的对plp1基因的编辑恢复了髓鞘相关障碍佩梅病(pmd)的jimpy小鼠模型的正常功能和完整寿命以及在体外pmd模型神经胶质细胞中恢复功能。不受限于理论,相信通过失活plp1基因绕开了plp1相关的毒性,从而增强了经修饰的神经胶质细胞或少突胶质细胞生产功能性髓鞘的能力。
本发明描述的是plp1基因或其调控元件中的位点特异性的插入或缺失(插入缺失)。当靶向plp1基因时,这将提供plp1表达的有效和永久的失活或降低。定点核酸酶或基因编辑plp1基因破坏组合物可例如使用以下来递送:一个或多个aav载体、整合酶缺陷型慢病毒载体(idlv)和/或核酸(诸如质粒、小环质粒和寡核苷酸)。
本发明中还描述了plp1基因或其基因调控元件的精确原位编辑以在神经干细胞(nsc)和/或少突胶质细胞前体细胞(opc)及其后代中产生增强的功能性髓鞘产生能力。核酸酶或基因编辑介导将破坏性突变原位引入nsc和/或opc中的内源性plp1基因,为编辑细胞的后代带来这些增强的能力。另外,plp1调控基因的特异性敲除或plp1调控基因原位突变到nsc或opc中的内源性基因中为这些细胞的后代赋予了强化/改进的功能性髓鞘产生。
可将所述细胞和方法移植到动物模型/或人类患者中而不会显著影响细胞功能和活性。此外,细胞保持其在体内持续存在的能力,并且能在移植后增强受试者的功能性髓鞘产生。
除非另外表明,本发明所公开的方法的实施以及组合物的制备和使用采用分子生物学、生物化学、染色质结构和分析、计算化学、细胞培养、重组dna和相关领域的常规技术,如同在该领域的技术范围内那样。这些技术在文献中有详细解释。参见,例如,sambrook等molecularcloning:alaboratorymanual,第二版,coldspringharborlaboratorypress,1989和第三版,2001;ausubel等,currentprotocolsinmolecularbiology,johnwiley&sons,newyork,1987和定期更新;methodsinenzymology系列,academicpress,sandiego;wolffe,chromatinstructureandfunction,第三版,academicpress,sandiego,1998;methodsinenzymology,vol.304,“chromatin”(p.m.wassarman和a.p.wolffe,eds.),academicpress,sandiego,1999;andmethodsinmolecularbiology,vol.119,“chromatinprotocols”(p.b.becker,ed.)humanapress,totowa,1999。
在以下部分说明本发明的其他方面。
融合分子
本发明描述了可用于切割细胞中选定的靶基因(例如,plp1或其基因调控元件)的组合物,例如,核酸酶。在一些实施方案中,融合分子(例如,核酸酶)的一个或多个组成部分是天然的。在其他实施方案中,融合分子(例如,核酸酶)的一个或多个组成部分是非天然的,即,在dna-结合结构域和/或切割结构域中经过工程化的。例如,天然核酸酶的dna-结合结构域可被改变为结合选定的靶位点(例如,已被工程化以结合不同于同源结合位点的位点的大范围核酸酶(meganuclease))。在其他实施方案中,核酸酶包括异源性dna-结合结构域和切割结构域(例如,锌指核酸酶;tal-效应子结构域dna结合蛋白;异源性切割结构域中的大范围核酸酶dna-结合结构域)。
dna-结合结构域
在一些实施方案中,本发明所述的组合物和方法采用结合供体分子和/或结合细胞基因组中的所关注的区域的大范围核酸酶(归位内切酶)dna-结合结构域。天然的大范围核酸酶识别15-40个碱基对的切割位点,并且通常分为四个家族:laglidadg家族,gly-ylg家族,his-cystbox家族,和hnh家族。示例性的归位内切酶包括i-scei、i-ceui、pi-pspi、pi-scei-sceiv、i-csmi、i-pani、i-sceii、i-ppoi、i-sceiii、i-crei、i-tevi、i-tevii和i-teviii。其识别序列是已知的。也参见美国专利5,420,032;美国专利6,833,252;belfort等(1997)nucleicacidsres.25:3379-3388;dujon等(1989)gene82:115-118;perler等(1994)nucleicacidsres.22,1125-1127;jasin(1996)trendsgenet.12:224-228;gimble等(1996)j.mol.biol.263:163-180;argast等(1998)j.mol.biol.280:345-353;以及newenglandbiolabs目录。此外,归位内切酶和大范围核酸酶的dna-结合特异性可被工程化以结合非天然靶位点。参见,例如,chevalier等(2002)molec.cell10:895-905;epinat等(2003)nucleicacidsres.31:2952-2962;ashworth等(2006)nature441:656-659;paques等(2007)currentgenetherapy7:49-66;美国专利公开号20070117128。归位内切酶和大范围核酸酶的dna-结合结构域可在核酸酶整体的背景下改变(即,使得核酸酶包括同源切割结构域),或可与异源性切割结构域融合。
在其他实施方案中,本发明所述方法和组合物中所用的一种或多种核酸酶的dna-结合结构域包括天然或工程化(非-天然)的tal效应子dna结合结构域。参见,例如,美国专利8,586,526,上述文献通过引用以其整体并入本发明。已知黄单胞菌属的植物病原菌在重要的作物植物中引起许多疾病。黄单胞菌的病原性取决于保守的iii型分泌(t3s)系统,其将多于25种不同的效应蛋白注入植物细胞中。在这些注入蛋白中包含转录活化因子样(tal)效应子,其模拟植物转录活化因子,并且操纵植物转录组(参见kay等(2007)science318:648-651)。这些蛋白质包含dna结合结构域和转录活化结构域。最有特色的tal-效应子之一是来自野油菜黄单胞菌辣椒斑点病致病变种(xanthomonascampestgrispv.vesicatoria)的avrbs3(参见bonas等(1989)molgengenet218:127-136和wo2010079430)。tal-效应子包含串联重复的集中结构域,每个重复包含约34个氨基酸,这是这些蛋白质的dna结合特异性的关键。另外,它们包含核定位序列和酸性转录活化结构域(参见schornacks,等(2006)jplantphysiol163(3):256-272)。另外,在植物病原菌青枯雷尔氏菌(ralstoniasolanacearum)中,已发现了两个被命名为brg11和hpx17的基因,它们与青枯菌生物变种1菌株gmi1000和生物变种4菌株rs1000中的黄单胞菌属的avrbs3家族同源(参见heueretat(2007)applandenvirmicro73(13):4379-4384)。这些基因的核苷酸序列彼此有98.9%相同,但区别在于在hpx17的重复结构域中的1,575bp缺失。然而,两种基因产物与黄单胞菌的avrbs家族的蛋白质的序列同一性均小于40%。参见,例如,美国专利8,586,526,上述文献通过引用以其整体并入本发明。
这些tal效应子的特异性取决于在串联重复中发现的序列。重复序列包括约102bp,并且重复通常彼此91-100%同源(bonas等,出处同前)。重复的多态性通常位于12和13位,并且在12和13位的高变双残基(rvd)的同一性与tal-效应子的靶序列中的连续核苷酸的同一性之间存在一一对应关系(参见moscouandbogdanove,(2009)science326:1501,andboch等(2009)science326:1509-1512)。通过实验,已经确定了这些tal-效应子的dna识别的天然代码,使得12和13位的hd序列导致与胞嘧啶(c)的结合,ng结合t,ni结合a、c、g或t,nn结合a或g,并且ing结合t。这些dna结合重复已组装成具有新的组合和数量的重复的蛋白质,以制造能够与新序列互相作用并激活植物细胞中非内源性报告基因的表达(boch等,出处同前)。工程化tal蛋白质已与fokl切割半结构域连接以产生tal效应子结构域核酸酶融合(talen)。参见,例如,美国专利8,586,526;christian等((2010)<geneticsepub10.1534/genetics.110.120717)。在一些实施方案中,tale结构域包括如美国专利8,586,526中所述的n-cap和/或c-cap。
在一些实施方案中,用于体内切割和/或靶向切割细胞基因组的一种或多种核酸酶的dna结合结构域包括锌指蛋白。优选地,锌指蛋白是非天然存在的,因为其被工程化以结合所选择的靶位点。参见,例如,beerli等(2002)naturebiotechnol.20:135-141;pabo等(2001)ann.rev.biochem.70:313-340;isalan等(2001)naturebiotechnol.19:656-660;segal等(2001)curr.opin.biotechnol.12:632-637;choo等(2000)curr.opin.struct.biol.10:411-416;美国专利6,453,242;6,534,261;6,599,692;6,503,717;6,689,558;7,030,215;6,794,136;7,067,317;7,262,054;7,070,934;7,361,635;7,253,273;以及美国专利公开号2005/0064474;2007/0218528;2005/0267061,上述所有文献通过引用以其整体并入本发明。
与天然锌指蛋白相比,工程化锌指结合结构域可具有新的结合特异性。工程化方法包括但不限于理性设计和多种类型的选择。理性设计包括例如使用包含三联体(或四联体)核苷酸序列和单个锌指氨基酸序列的数据库,其中每个三联体或四联体核苷酸序列与结合特定三联体或四联体序列的锌指的一个或多个氨基酸序列相连。参见,例如,共同拥有的美国专利6,453,242和6,534,261,上述文献通过引用以其整体并入本发明。
示例性的选择方法包括噬菌体展示和双杂交系统,其在以下文献中公开:美国专利5,789,538;5,925,523;6,007,988;6,013,453;6,410,248;6,140,466;6,200,759;和6,242,568;以及wo98/37186;wo98/53057;wo00/27878;wo01/88197和gb2,338,237。另外,已在例如共同拥有的wo02/077227中描述了锌指结合结构域的增强的结合特异性。
另外,如这些和其他参考文献中所公开的,锌指结构域和/或多指锌指蛋白可使用任何合适的连接序列连接在一起,包括,例如,长度为5个或更多个氨基酸的连接序列。也参见美国专利6,479,626;6,903,185;和7,153,949中长度为6个或更多个氨基酸的示例性连接序列。本发明所述的蛋白质包括蛋白质的各个锌指之间的合适连接序列的任何组合。
在一些方面,dna-结合结构域靶向plp1基因或plp1基因调控元件。
靶位点的选择;zfp以及融合蛋白(及编码其的多核苷酸)的设计和构建是本领域技术人员已知的,并且在以下中详细描述:美国专利6,140,081;5,789,538;6,453,242;6,534,261;5,925,523;6,007,988;6,013,453;6,200,759;wo95/19431;wo96/06166;wo98/53057;wo98/54311;wo00/27878;wo01/60970wo01/88197;wo02/099084;wo98/53058;wo98/53059;wo98/53060;wo02/016536;和wo03/016496。
另外,如这些和其他参考文献中所公开的,锌指结构域和/或多指锌指蛋白可使用任何合适的连接序列连接在一起,包括,例如,长度为5个或更多个氨基酸的连接序列。也参见美国专利6,479,626;6,903,185;和7,153,949中长度为6个或更多个氨基酸的示例性连接序列。本发明所述的蛋白质包括蛋白质的各个锌指之间的合适连接序列的任何组合。
在一些实施方案中,dna-结合结构域是crispr/cas核酸酶系统的一部分。参见,例如,美国专利8,697,359和美国专利申请序列号14/278,903。编码系统的rna组成部分的crispr(成簇的规律间隔的短回文重复序列)基因座和编码蛋白质的cas(crispr-相关的)基因座(jansen等,2002.mol.microbiol.43:1565-1575;makarova等,2002.nucleicacidsres.30:482-496;makarova等,2006.biol.direct1:7;haft等,2005.ploscomput.biol.1:e60)构成了crispr/cas核酸酶系统的基因序列。微生物宿主的crispr基因座包含crispr-相关(cas)基因以及能够调控crispr-介导的核酸切割的特异性的非编码rna元件的组合。crispr-cas系统被分为两类。第1类使用数个cas蛋白与crisprrna(crrna)共同建立功能性核酸内切酶。第2类crispr系统使用单个cas蛋白以及crrna。
第2类ii型crispr是最有特色的系统之一,并且在四个连续步骤中实施靶向dna双链断裂。首先从crispr基因座转录两种非-编码rna,前体-crrna阵列和tracrrna。其次,tracrrna与前体-crrna的重复区杂交并介导将前体-crrna加工成包含单个间隔序列的成熟crrna。第三,成熟crrna:tracrrna复合物通过crrna上的间隔序列与邻近前间区序列邻近基序(pam)(靶向识别的额外要求)的靶dna上的前间区序列之间的waston-crick碱基配对将功能域(例如,核酸酶诸如cas)导向靶dna。最后,cas9介导切割靶dna以在前间区序列内产生双链断裂。crispr/cas系统的活性包括三个步骤:(i)在被称为“适应”的过程中将外来dna序列插入到crispr阵列中以防止未来的攻击,(ii)表达相关蛋白质,以及表达和加工阵列,随后是(iii)rna-介导的与外来核酸的相互作用。因此,在细菌细胞中,数种所谓的‘cas’蛋白与crispr/cas系统的天然功能相关,并且在诸如插入外来dna等的功能中发挥作用。
在一些实施方案中,cas蛋白可以是天然“cas”蛋白的“功能性衍生物”。天然序列多肽的“功能性衍生物”是具有与天然序列多肽共同的定性生物特性的化合物。“功能性衍生物”包括但不限于天然序列的片段以及天然序列多肽衍生物及其片段,条件是其具有与相应的天然序列多肽共同的生物活性。本发明所设想的生物活性是功能性衍生物将dna底物水解为片段的能力。术语“衍生物”包括多肽的氨基酸序列变体、其共价修饰和融合,诸如衍生的cas蛋白。cas多肽或其片段的合适衍生物包括但不限于cas蛋白或其片段的突变体、融合体、共价修饰。cas蛋白包括cas蛋白或其片段以及cas蛋白或其片段的衍生物,其可得自细胞或者化学合成或者这两种程序的组合。细胞可以是天然产生cas蛋白的细胞,或者天然产生cas蛋白并且被遗传工程化以便以较高表达水平产生内源性cas蛋白或产生来自外源引入核酸的cas蛋白的细胞,所述核酸编码相同或不同于内源性cas的cas。在一些情况中,细胞不会天然产生cas蛋白,并且被遗传工程化以产生cas蛋白。在一些实施方案中,cas蛋白是用于通过aav载体递送的小cas9直系同源物(ran等(2015)nature510,p.186)。在特定实施方案中,cas蛋白是sacas9或spcas9cas蛋白。
在一些实施方案中,cas9蛋白是哺乳动物cas9或来自以下的cas9:化脓性链球菌(streptococcuspyogenes)、脑膜炎双球菌(neisseriameningitidis)、嗜热链球菌(streptococcusthermophilus)、肺炎链球菌(streptococcuspneumnoniae)、结肠弯曲菌(campylobactercoli)、空肠弯曲菌(campylobacterjejuni)、变异链球菌(streptococcusmutans)、多杀性巴氏杆菌(pasteurellamultocida)、长双歧杆菌(bifidobacteriumlongum)、史氏芽孢杆菌(bacillussmithii)、齿密螺旋体(treponemadenticola)、犬支原体(mycoplasmacanis)或粪肠球菌(enterococcusfaecalis)、华德萨特菌(sutterellawadsworthensis)、filifactoralocis、约氏乳杆菌(lactobacillusjohnsonii)、红嘴鸥弯曲杆菌(campylobacterlari)、白喉杆菌(corynebacterdiptheriae)、parvibaculumlavamentivorans、鸡霉形体(mycoplasmagallisepticum)、金黄色葡萄球菌亚种金黄色葡萄球菌(staphylococcusaureussubsubspeciesaureus)、巴黎嗜肺军团菌(legionellapneumophilaparis)、齿密螺旋体(treponemadenticola)、假中间葡萄球菌(staphylococcuspseudintermedius)或灰色奈瑟球菌(neisseriacinerea)。
在一些实施方案中,cas9蛋白具有一个或多个核定位序列。
在一些实施方案中,dna-结合结构域是第2类v型crispr/cascpf1系统的一部分。像cas9一样,cpf1核酸酶包含ruvc-样核酸内切酶结构域,但是它们缺乏cas9的第二hnh核酸内切酶结构域和n-末端α-螺旋识别瓣叶。cpf1以交错模式切割dna,并且仅需要一个rna,而不像cas9切割时所需的两个(tracrrna和crrna)。cpf1的交错切割模式开启了定向基因转移的可能性,类似于传统的限制性内切酶克隆。粘性末端介导的基因转移可特别有助于靶向难以通过同源定向修复(hdr)修饰的非分裂细胞。cpf1还扩展了能够通过crispr靶向富at区域或富at基因组的位点的数量,所述富at区域或富at基因组缺乏spcas9所支持的3’-nggpam位点(参见,例如,zetsche等(2015)cell163(3):759-771和makarova等,(2015)naturereviewsmicrobiology13(11):722-736)。
在一些实施方案中,dna结合结构域是适合用于人细胞基因组编辑的argonaute核酸内切酶系统的一部分。适合用于人细胞基因组编辑的示例性的argonaute核酸内切酶系统可包括来自格氏嗜盐碱杆菌(natronobacteriumgregoryi)的argonautedna-引导的核酸内切酶(ngago)(gao等(2016)naturebiotechnology.34:768-773)。ngago结合~24个核苷酸的5′磷酸化单链向导dna(gdna),并且在加载了gdna时有效地产生位点特异性dna双链断裂。使用5’磷酸化ssdna作为向导分子减少了细胞寡核苷酸误导ngago的可能性。向导分子仅可在蛋白表达期间连接到ngago。一旦加载了向导,则ngago不能交换自由浮动ssdna的gdna。ngago–gdna系统不像cas9那样需要前间区序列邻近基序(pam),并且显示对向导-靶错配的低容忍性和在编辑富(g+c)的基因组靶中的高效。
任意dna序列的外源向导dnas可被加载到ngago蛋白上。由于ngago切割的特异性由向导dna指导,因此与外源的、研究者指定的向导dna形成的ngago-dna复合物将会把ngago靶向dna切割导向互补的研究者指定的靶dna。以此方式,可在dna中产生靶向双链断裂。使用ngago-向导dna系统(或来自其他生物体的直系同源ago-向导dna系统)允许细胞内基因组dna的靶向切割。这样的切割可以是单链或双链的。对于哺乳动物基因组dna的切割,将优选使用针对哺乳动物细胞中表达进行优化的ngago密码子形式。此外,可优选用在体外形成的ngago-dna复合物处理细胞,其中ngago蛋白与细胞渗透肽融合。此外,可优选使用已通过诱变改变为在37℃时具有改进的活性的ngago蛋白形式。ago-rna-介导的dna切割可用于使用dna断裂研究领域中的标准技术来影响包括基因敲除、靶基因加成、基因纠正、靶基因缺失的结果的多样性。
因此,核酸酶包括特异性结合任何基因(例如,plp1)中的靶位点的dna-结合结构域,其中希望向所述基因中插入或缺失碱基(产生插入缺失)以失活或降低基因表达。
切割结构域
任何合适的切割结构域都可被可操作地连接到dna-结合结构域以形成核酸酶。例如,zfpdna-结合结构域与核酸酶结构域融合以产生zfn-一种能够通过其工程化的(zfp)dna结合结构域识别其预期核酸靶并通过核酸酶活性使得dna在zfp结合位点附近切割的功能性实体,包括用于多种生物体的基因组修饰。参见,例如,美国专利公开20030232410;20050208489;20050026157;20050064474;20060188987;20060063231;和国际公开wo07/014275。类似地,taledna-结合结构域与核酸酶结构域融合以产生talen。参见,例如,美国专利8.586,526。
如上所述,切割结构域可与dna-结合结构域异源,例如锌指dna-结合结构域和来自核酸酶的切割结构域或talendna-结合结构域以及来自不同核酸酶的切割结构域或大范围核酸酶dna-结合结构域和切割结构域。异源性切割结构域可由任何核酸内切酶或核酸外切酶获得。可作为切割结构域来源的示例性的核酸内切酶包括但不限于限制性核酸内切酶和归位核酸内切酶。切割dna的其他酶是已知的(例如,s1核酸酶;绿豆核酸酶;胰脱氧核糖核酸酶i;微球菌核酸酶;酵母ho核酸内切酶)。一种或多种这些酶(或其功能片段)可用作切割结构域和切割半结构域的来源。
类似地,如上所述,切割半结构域可来源于任何核酸酶或其部分,其需要二聚以获得切割活性。通常,如果融合蛋白包含切割半结构域,则需要两个融合蛋白来切割。或者可选地,可使用包含两个切割半结构域的单个蛋白质。两个切割半结构域可来源于相同的核酸内切酶(或其功能片段),或者每个切割半结构域可来源于不同的核酸内切酶(或其功能片段)。另外,两个融合蛋白的靶位点优选相对于彼此布置,使得两个融合蛋白与其各自的靶位点的结合将切割半结构域布置为彼此空间定位,这允许切割半结构域形成功能性切割结构域,例如,通过二聚。因此,在一些实施方案中,靶位点的相近边缘通过5-8个核苷酸或通过15-18个核苷酸隔开。然而在两个靶位点之间可插入任何整数个核苷酸或核苷酸对(例如,2-50个核苷酸对或更多)。通常,切割位点位于靶位点之间。
限制性核酸内切酶(限制性内切酶)存在于许多物种中,并且能够与dna序列特异性结合(在识别位点处)和在结合位点处或附近切割dna。一些限制性内切酶(例如,iis型)在远离识别位点处的位点切割dna,并且具有可分离的结合结构域和切割结构域。例如,iis型酶foki在一条链上在距其识别位点9个核苷酸处和在另一条链上距离其识别位点13个核苷酸处催化dna的双链切割。参见,例如,美国专利5,356,802;5,436,150和5,487,994;以及li等(1992)proc.natl.acad.sci.usa89:4275-4279;li等(1993)proc.natl.acad.sci.usa90:2764-2768;kim等(1994a)proc.natl.acad.sci.usa91:883-887;kim等(1994b)j.biol.chem.269:31,978-31,982。因此,在一个实施方案中,融合蛋白包含来自至少一个iis型限制性内切酶的切割结构域(或切割半结构域)和一个或多个锌指结合结构域,其可经过或未经过工程化。
其切割结构域可与结合结构域分离的示例性的iis型限制性内切酶是foki。这一特定酶作为二聚体是有活性的。bitinaite等(1998)proc.natl.acad.sci.usa95:10,570-10,575。因此,对于本发明的目的,在所公开的融合蛋白中使用的foki酶的部分被视为切割半结构域。因此,对于使用锌指-foki融合体的细胞序列的靶向双链切割和/或靶向替换,可使用各自包含foki切割半结构域的两个融合蛋白以重构具有催化活性的切割结构域。或者可选地,也可使用包含锌指结合结构域和两个foki切割半结构域的单个多肽分子。使用锌指-foki融合体的靶向切割和靶向序列改变的参数在本发明中其他地方提供。
切割结构域或切割半结构域可为保持切割活性或保持多聚(例如二聚)以形成功能性切割结构域的能力的蛋白质的任何部分。
示例性的iis型限制性内切酶在国际公布wo07/014275中描述,上述文献整体并入本发明。其他限制性内切酶还包含可分离的结合结构域和切割结构域,这些均被本发明所设想。参见,例如,roberts等(2003)nucleicacidsres.31:418-420。
在一些实施方案中,切割结构域包含一个或多个工程化切割半结构域(也称为二聚结构域突变体),其最小化或阻止均二聚化,如例如以下中所述的:美国专利公开号20050064474;20060188987;20070305346和20080131962,上述所有文献的公开内容通过引用以其整体并入本发明。fok1的446、447、479、483、484、486、487、490、491、496、498、499、500、531、534、537和538位的氨基酸残基都是影响fok1切割半结构域的二聚的靶点。
可使用具有多于一个突变的切割结构域,例如在一个切割半结构域的490位(e→k)和538位(i→k)突变以产生名为“e490k:1538k"的工程化切割半结构域,并且在另一个切割半结构域的486位(q→e)和499位(i→l)突变以产生名为“q486e:i499l的工程化切割半结构域”;在486位用glu(e)残基替换野生型gln(q)残基、在499位用leu(l)残基替换野生型iso(i)残基和在496位用asp(d)或glu(e)残基替换野生型asn(n)残基的突变(也分别称为“eld”和“ele”结构域);包含在490、538和537位(相对于野生型foki编号)的突变的工程化切割半结构域,例如在490位用lys(k)残基替换野生型glu(e)残基、在538位用lys(k)残基替换野生型iso(i)残基和在537位用lys(k)残基或arg(r)残基替换野生型his(h)残基的突变(也分别称为“kkk”和“kkr”结构域);和/或包含在490和537位(相对于野生型foki编号)的突变的工程化切割半结构域,例如在490位用lys(k)残基替换野生型glu(e)残基和在537位用lys(k)残基或arg(r)残基替换野生型his(h)残基的突变(也分别称为“kik”和“kir”结构域)。参见,例如,美国专利7,914,796;8,034,598和8,623,618,其公开内容通过引用以其整体并入用于所有目的。在其他实施方案中,工程化切割半结构域包括“sharkey”和/或“sharkey’“突变(参见guo等,(2010)j.mol.biol.400(1):96-107)。
或者可选地,可使用所谓的“分解酶(split-enzyme)”技术在核酸靶位点在体内组装核酸酶(参见,例如,美国专利公开号20090068164)。这种分解酶的组成部分可在单独的表达构建物上表达,或者可连接在一个开放阅读框内,在这里各个组成部分通过例如自切割2a肽或ires序列间隔开。组成部分可以是单个锌指结合结构域或大范围核酸酶核酸结合结构域的结构域。
在使用前可对核酸酶的活性进行筛选,例如,在如美国专利8,563,314中所述的酵母基染色体系统。
cas9相关的crispr/cas系统包含两个rna非-编码组成部分:tracrrna和包含由相同的直接重复(dr)隔开的核酸酶向导序列(间隔子)的前体-crrna阵列。为使用crispr/cas系统实现基因组工程化,这些rna的两项功能都必须存在(参见cong等,(2013)sciencexpress1/10.1126/science1231143)。在一些实施方案中,tracrrna和前体-crrna经由单独的表达构建物或作为单独的rna供应。
在其他实施方案中,构建嵌合rna,其中将工程化成熟crrna(赋予靶特异性)与tracrrna(提供与cas9的相互作用)融合以产生嵌合的cr-rna-tracrrna杂合体(也称为单一向导rna或sgrna)。因此,与cas9核酸酶一起,本发明方法中对plp1基因的修饰需要引入含有对不可变的支架序列的靶dna5’有特异性的约20个碱基序列的sgrna。sgrna可作为rna递送,或通过在启动子下用具有sgrna-编码序列的质粒转化来递送。在特定实施方案中,用于修饰plp1基因的sgrna序列包括但不限于靶向plp1基因的外显子3的aagaccaccatctgcggcaangg(seqidno:1)和ccagcaggagggccccataangg(seqidno:2),靶向plp1基因的外显子1的gtcagagtgccaaagacatggnngrrt(seqidno:3)。
靶位点
如上文中所详述地,dna-结合结构域可被工程化以结合所选择的任何序列。工程化的dna-结合结构域与天然的dna-结合结构域相比可具有新的结合特异性。
在一些实施方案中,靶位点是内源性plp1基因的外显子。用于靶定的合适的基因组外显子区域的非限制性实例包括plp1的外显子1或3或7。在一些实施方案中,靶位点包括plp1的外显子3的5’上的约80个核苷酸片段。
在一些实施方案中,核酸酶靶向plp1基因。在一些实施方案中,核酸酶靶向plp1基因调控元件如启动子或增强子。
供体
在一些实施方案中,本发明涉及由核酸酶-介导的外源基因靶向整合到细胞基因组中。如上所述,插入外源序列(也称为“供体序列”或“供体”或“转基因”),例如用于删除指定区域(诸如删除plp1基因复制体的一个拷贝,其出现在约70%的pmd患者中)和/或纠正突变基因(诸如plp1点突变,其出现在约30%的pmd患者中)或用于提高野生型基因的表达。显而易见的是,供体序列通常不同于其所放置的基因组序列。供体序列可包含两侧是两个同源区域的非-同源序列以允许在所关注的位置的有效hdr,或者可通过非同源定点修复机制整合。另外,供体序列可包括包含与细胞染色质中的所关注的区域不同源的序列的载体分子。供体分子可包含若干个与细胞dna同源的不连续区域。此外,为靶向插入通常不存在于所关注的区域中的序列,所述序列可存在于供体核酸分子中,并且两侧是与所关注的区域中的序列同源的区域。
与核酸酶一样,可将供体引入任何形式。在一些实施方案中,可通过本领域已知方法使用dna和/或病毒载体引入供体。参见,例如,美国专利公开号20100047805和20110207221。可以双链或单链形式引入供体。可以环状或线性形式引入供体。如果以线性形式引入,可通过本领域技术人员已知的方法对供体序列的末端进行保护(例如,保护免于核酸外切降解)。例如,可将一个或多个双脱氧核苷酸残基添加到线性分子的3′末端和/或将自身互补的寡核苷酸连接到一个或两个末端。参见,例如,chang等(1987)proc.natl.acad.sci.usa84:4959-4963;nehls等(1996)science272:886-889。保护外源多核苷酸免于降解的其他方法包括但不限于添加末端氨基和使用修饰的核苷酸间键,诸如,例如,硫代磷酸酯、氨基磷酸酯和o-甲基核糖或脱氧核糖残基。
在一些实施方案中,供体包括长度大于1kb的序列(例如,编码序列,也称为转基因),例如2-200kb,2-10kb(或其间的任何值)。供体还可包括至少一个核酸酶靶位点。在一些实施方案中,供体包括至少2个靶位点,例如用于zfn、talen、ngago或crispr/cas核酸酶对。通常,核酸酶靶位点位于转基因序列之外,例如,转基因序列的5′和/或3′,以切割转基因。核酸酶切割位点可用于任何核酸酶。在一些实施方案中,双链供体中所含的核酸酶靶位点是用于切割内源性靶的相同核酸酶,其中切割供体经由非同源依赖性方法整合到所述内源性靶中。
可插入供体以使得通过整合位点处的内源性启动子驱动其表达,即,驱动插入供体的内源性基因表达的启动子。然而,显而易见的是,供体可包含启动子和/或增强子,例如组成型启动子或可诱导或组织特异性启动子。
可将供体分子插入内源性基因中以表达全部或一些内源性基因或不表达内源性基因。在一些实施方案中,将转基因整合到plp1或plp1基因调控元件中以失活plp1或降低其表达。
此外,尽管不是表达所必需的,但外源序列还可包括转录调控或翻译调控或其它序列,例如,启动子、增强子、绝缘子、内部核糖体进入位点、编码2a肽的序列和/或多腺苷酸化信号。另外,可包含剪接受体序列。示例性的剪接受体位点序列是本领域技术人员已知的,仅通过举例的方式,其包含ctgacctcttctcttcctcccacag(seqidno:4)(来自人hbb基因)和tttctctccacag(seqidno:5)(来自人免疫球蛋白-γ基因)。
可使用本领域已知的标准技术如pcr将本发明所述的供体序列(转基因和/或修复模板)从质粒、细胞或其它来源分离出来。所用的供体可包括多种类型的拓扑学,包括环状超螺旋、环状松弛型、线性等。或者可选地,其可使用标准寡核苷酸合成技术来化学合成。另外,供体可为甲基化或非甲基化的。供体可为细菌或酵母人工染色体形式(bac或yac)。
本发明所述的供体多核苷酸可包括一个或多个非-天然碱基和/或主链。特别是,可使用本发明所述方法进行带有甲基化胞嘧啶的供体分子的插入以实现所关注的区域的转录静止状态。
外源(供体)多核苷酸可包含任何所关注的序列(外源序列)。示例性的外源序列包括但不限于任何多肽编码序列(例如,cdna)、启动子序列、增强子序列、表位标签、标记基因、切割酶识别位点和多重类型的表达构建物。标记基因包括但不限于:编码介导抗生素抗性(如阿莫西林抗性、新霉素抗性、g418抗性、嘌呤霉素抗性)的蛋白质的序列,编码彩色或荧光或发光蛋白(例如,绿色荧光蛋白、增强绿色荧光蛋白、红色荧光蛋白、荧光素酶)和介导增强细胞生长和/或基因扩增的蛋白(例如,二氢叶酸还原酶)的序列。表位标签包括,例如,flag、his、myc、tap、ha或任何可检测氨基酸序列的一个或多个拷贝。
在一些实施方案中,供体进一步包含编码需要在细胞中表达的任何多肽的多核苷酸,所述需要在细胞中表达的多肽包括但不限于抗体、抗原、酶、受体(细胞表面或核)、激素、淋巴因子、细胞因子、报告多肽、生长因子以及上述任何的功能片段。编码序列可为,例如,cdna。
在一些实施方案中,外源序列可包括允许选择已经历靶向整合的细胞的标记基因(如上所述)和编码其它功能性的连接序列。标记基因的非限制性实例包括gfp、药物选择标记等。
在一些实施方案中,供体可包括,例如,替换突变的有害内源性序列的野生型基因。例如,可向其中基因的内源性拷贝发生突变的干细胞基因组中插入野生型(或其它功能性)基因序列。在其他实施方案中,供体可包括,例如,替换野生型内源性基因的突变基因。例如,可向干细胞基因组中插入野生型plp1序列以突变髓鞘相关障碍中涉及的内源性有害突变plp1基因。
遵循本发明的教导构建这样的表达盒,其利用了分子生物学领域中公知的方法(参见,例如,ausubel或maniatis)。在使用表达盒产生转基因动物之前,可通过将表达盒引入合适的细胞系(例如,原始细胞、转化细胞或永生化细胞系)来测试表达盒对于与选定控制元件相关的应激诱导剂的响应性。
此外,尽管不是表达所必须的,外源序列还可包括转录或翻译调控序列,例如,启动子、增强子、绝缘子、内部核糖体进入位点、编码2a肽的序列和/或多腺苷酸化信号。此外,所关注的基因的控制元件能够可操作地连接报告基因以产生嵌合基因(例如,报告基因表达盒)。
也可实现非-编码核酸序列的靶向插入。编码反义rna、rnai、sirna、shrna和微小rna(mirna)的序列也可用于靶向插入。
在其它实施方案中,供体核酸可包含作为其他核苷酸设计的特异性靶点的非-编码序列。随后,可在细胞中表达其它核酸酶以切割原始供体分子并通过插入另一种所关注的供体分子进行修饰。以此方式,可产生供体分子的反复整合,使得在特定的所关注的基因座或在安全港基因座进行特征叠加。
在一些实施方案中,可使用在佩梅病(pmd)的jimpy小鼠模型中评估髓鞘再生和临床严重性降低的体内测定方法进一步筛选本发明所述的治疗性基因修饰组合物。
基因沉默
在一些实施方案中,核酸酶靶向plp1基因。在一些实施方案中,核酸酶靶向plp1基因调控元件诸如启动子或增强子。
在一些实施方案中,对内源性plp1基因产物或促进plp1表达的plp1调控元件的基因产物进行修饰以降低plp1表达水平。在一些实施方案中,使用基因沉默对内源性plp1基因产物或plp1调控元件的基因产物进行修饰以降低细胞中的plp1表达水平。
在其他实施方案中,通过在有此需要的受试者的组织或细胞中使用减少或抑制plp1或促进plp1表达的plp1调控元件的表达的基因沉默剂对内源性plp1基因产物或plp1调控元件的基因产物进行修饰。“表达”是指从基因产生基因产物(通常为蛋白质(任选地,在翻译后修饰)或功能/结构rna)的信息总体流动。
在一些实施方案中,试剂可包括抑制或降低细胞中的plp1表达的rnai构建物。rnai构建物包括可特异性阻断靶基因表达的双链rna。“rna干扰”或“rnai”是最初应用于在植物和蠕虫中观察到的双链rna(dsrna)以特异性和转录后方式阻断基因表达的现象的术语。
本发明所用的术语“dsrna”是指包含双链特征并且能够在细胞中加工成sirna的sirna分子或其它rna分子,诸如发夹rna部分(shrna)和微小-rna(mirna)。
当术语“功能丢失”涉及受主题rnai方法抑制的基因时,其是指与不存在rnai构建物时的水平相比基因的表达水平降低。
如本发明所用的,短语“介导rnai”是指(表示)区分哪些rna会通过rnai过程降解的能力,例如,以序列特异性方式而不是通过序列依赖性dsrna反应(例如,pkr反应)发生降解。
本发明所用的术语“rnai构建物”在整个说明书中使用的通用术语,其包括小干扰rna(sirna)、发夹rna和其它rna种类(诸如mirna),其可在体内切割而形成sirna和/或mirna。本发明的rnai构建物还包括能够产生在细胞中形成dsrna或发夹rna的转录物和/或能在体内产生sirna的转录物的表达载体。
“rnai表达载体”(在本发明中也称为“dsrna-编码质粒”)是指用于表达(转录)rna的可复制核酸构建物,其在表达该构建物的细胞中产生sirna/shrna/mirna。这样的载体包括转录单元,所述转录单元包含以下的组装体:(1)在基因表达中具有调控作用的基因元件,例如,启动子、操纵子或增强子,其可操作地连接到(2)转录产生双链rna(在细胞中退火形成sirna或可被加工为sirna或mirna的单个发夹rna的两个rna部分)的“编码”序列,和(3)合适的转录起始和终止序列。
启动子和其它调控元件的选择通常根据预期宿主细胞而不同,诸如opc。通常,可用于重组dna技术中的表达载体通常为“质粒”形式,其是指环状的双链dna环,其载体形式不与染色体结合。在本说明书中,“质粒”和“载体”可互换使用,因为质粒是最常用的载体形式。然而,本申请描述了具有等同功能的其它形式的表达载体,在此之后其为本领域所知。
rnai构建物包含在细胞生理条件下与要被抑制的基因(即,“靶”基因)的mrna转录物的至少一部分的核苷酸序列杂交的核苷酸序列。双链rna仅需与具有介导rnai能力的天然rna足够相似即可。因此,实施方案容忍可能因基因突变、菌株多样性或进化趋异性而可预期的序列变异。可容忍的靶序列与rnai构建物序列之间的核苷酸错配数目不多于:5个碱基对中有1个,或者10个碱基对中有1个,或者20个碱基对中有1个,或者50个碱基对中有1个。sirna双链体中心的错配是最关键的,并且可以基本上消除靶rna的切割。相反,与靶rna互补的sirna链的3’末端的核苷酸对靶识别特异性没有特别影响。
可通过本领域已知的序列比较和比对算法来优化序列同一性,并且通过例如使用默认参数的bestfit软件程序中实施smith-waterman算法来计算核苷酸序列的百分比差异(例如,universityofwisconsingeneticcomputinggroup)。优选在抑制rna和靶基因部分之间有大于90%的序列同一性或甚至100%的序列同一性。或者可选地,rna的双链区域可在功能上定义为能够与靶基因转录物的一部分杂交的核苷酸序列。
可通过化学合成方法或通过重组核酸技术来进行rnai构建物的生产。所处理的细胞的内源性rna聚合酶可在体内介导转录,或者克隆的rna聚合酶可用于体外转录。rnai构建物可包括对磷酸-糖主链或核苷进行修饰,例如,以减少对细胞核酸酶的易感性、提高生物可利用度、改善制剂特性和/或改变其他药代动力学特性。例如,天然rna的磷酸二酯键可被修饰为包含氮或硫杂原子中的至少一种。可调整rna结构中的修饰以允许特异性基因抑制,同时避免对dsrna的一般响应。类似地,可修饰碱基以阻断腺苷脱氨酶的活性。rnai构建物可酶促生产或通过部分/完全的有机合成生产,可通过体外酶促或有机合成引入修饰的核糖核苷酸。
化学修饰rna分子的方法可适用于修饰rnai构建物(参见例如,nucleicacidsres,25:776-780;jmolrecog7:89-98;nucleicacidsres23:2661-2668;antisensenucleicaciddrugdev7:55-61)。仅举例说明,rnai构建物的主链可用以下来修饰:硫代磷酸酯、氨基磷酸酯、二硫代磷酸酯、嵌合的甲基磷酸酯-磷酸二酯、肽核酸、含有5-丙炔基-嘧啶的寡聚物或糖修饰(例如,2’-取代核糖核苷,a-构型)。
可通过单个自身互补的rna链或两个互补的rna链形成双链结构。rna双链体形成可始于细胞内部或外部。可以允许递送每个细胞至少一个拷贝的量引入rna。较多量(例如,至少5、10、100、500或1000个拷贝/细胞)双链材料可得到更有效的抑制,而较少量也可用于特定应用。抑制是序列特异性的,因为对应于rna双链体区域的核苷酸序列对于基因抑制是靶向的。
在一些实施方案中,主题rnai构建物是“小干扰rna”或“sirna”。这些核酸的长度为19-30个核苷酸左右,甚至更优选长度为21-23个核苷酸,例如,在长度上对应于核酸酶“切割”较长的双链rna所产生的片段。sirna被理解为募集核酸酶复合物并通过与特定序列配对将复合物引导至靶mrna。结果,蛋白复合物中的核酸酶将靶mrna降解。在特定实施方案中,21-23个核苷酸的sirna分子包含3’羟基。
可使用本领域技术人员已知的多种技术获得本发明所述的sirna分子。例如,可使用本领域已知的方法化学合成或重组产生sirna。例如,可合成短义和反义rna寡聚物并将其退火以形成在每端具有2-核苷酸突出端的双链rna结构(procnatlacadsciusa,98:742-9747;emboj,20:6877-88)。随后可通过被动摄入或所选择的递送系统(诸如如下所述者)将这些双链sirna结构直接引入细胞中。
在一些实施方案中,可通过加工较长的双链rna来生成sirna构建物,例如,在酶切酶存在下。在一个实施方案中,使用果蝇体外系统。在该实施方案中,将dsrna与来源于果蝇胚胎的可溶性提取物相结合,从而产生组合。将所述组合保持在如下条件下:其中将dsrna加工为约21至约23个核苷酸的rna分子。
可使用本领域技术人员已知的多种技术纯化sirna分子。例如,可使用凝胶电泳纯化sirna。或者可选地,可使用非变性方法(诸如非变性柱色谱)纯化sirna。另外,可使用色谱法(例如,尺寸排阻色谱法)、甘油梯度离心、抗体亲和纯化来纯化sirna。
在一些实施方案中,rnai构建物为发夹结构形式(名为发夹rna)。可外源合成发夹rna,或者可通过在体内从rna聚合酶iii启动子转录来形成发夹rna。在以下中描述了制备这样的发夹rna和将其在哺乳动物细胞中用于基因沉默的用途的实例:例如,genesdev,2002,16:948-58;nature,2002,418:38-9;rna,2002,8:842-50;和procnatlacadsci,2002,99:6047-52。优选地,这样的发夹rna在细胞或动物中进行工程化以确保对所需基因的连续和稳定的抑制。在本领域中已知可通过在细胞中加工发夹rna来生产sirna。
在一些实施方案中,rnai构建物为微小-rna或mirna的形式。mirna是通过rna聚合酶ii从其自身基因或从内含子转录的非-编码单链rna。在转录之后,首先将初级mirna加工为具有茎环结构的前体-mirna(约70个核苷酸),随后加工为功能性mirna(21-23个核苷酸)。与sirna和shrna一样,mirna也使用risc进行mrna降解和转录后基因沉默,并且具有靶向任何所关注的mrna的可能性。不同于与靶mrna完美互补的sirna,mirna不完美地结合靶mrna。这种部分互补结合允许每个mirna有可能与许多相似组的靶mrna相互作用。除了mrna降解之外,mirna可在没有内切核苷酸切割的情况下导致翻译抑制。因此mirna-靶基因可进行翻译调控而不会影响靶的mrna水平。mirna相对于sirna的优势是一个单一mirna转录物可被加工成多个sirna。
在其它的实施方案中,使用质粒递送双链rna,例如,作为转录产物。在这样的实施方案中,将质粒设计为包含rnai构建物的每一个有义链和反义链的“编码序列”。编码序列可为相同序列,例如,侧翼为反向启动子,或者可以是各自在单独启动子的转录控制下的单独序列。在转录了编码序列之后,互补的rna转录物碱基配对形成双链rna。
pct申请wo01/77350描述了用于转基因的双向转录以在真核细胞中获得同一转基因的有义和反义rna转录物的载体的实例。因此,一些实施方案提供了具有以下独特特性的重组载体:其包括具有两个重叠转录单元的病毒复制子,所述两个重叠转录单元以相反方向排列并且位于所关注的rnai构建物的转基因侧翼,其中这两个重叠转录单元从宿主细胞中的同一转基因片段同时得到有义和反义rna转录物。
在一些实施方案中,可使用慢病毒载体进行sirna的长期表达,诸如短-发夹rna(shrna),以敲减plp1基因的表达。虽然对于在基因治疗中使用慢病毒载体存在一些安全问题,但自失活慢病毒载体被认为是基因治疗的良好候选物,因为其容易转染哺乳动物细胞。
举例来说,可使用oligoengene软件(oligoengine,seattle,wa)产生plp1表达的短-发夹rna(shrna)下调,以鉴定作为sirna靶标的序列。可将寡核苷酸序列退火并连接到线性化psuperrnai载体(oligoengine,seattle,wa),并在大肠杆菌菌株dh5α细胞中转化。在选择阳性克隆之后,通过钙沉淀将质粒转染到293t细胞中。随后可使用所收集的包含shrna的病毒上清液感染哺乳动物细胞以下调plp1基因产物,从而降低细胞中的plp1表达水平。
还将可开发用于dna的载体用于rnai,这可部分归因于其相似的物理化学性质。这些载体大体上可分为两类:即,病毒和非病毒。参见对关于rnai的病毒递送系统的最新综述(castanotto和rossi,thepromisesandpitfallsofrna-interference-basedtherapeutics.nature457(7228):426–33,2009,其通过引用并入本发明)。非-病毒的rnai载体通常涉及将rnai构建物与带正电荷的载体(例如,阳离子细胞穿透肽、阳离子聚合物和树枝状大分子以及阳离子脂质)复合;将rnai构建物与小分子(例如,胆固醇、胆汁酸和脂质)、聚合物、抗体和rna缀合;以及将rnai构建物密封在纳米粒子制剂中。对rna主链进行修饰改善了sirna的稳定性而不会影响其rnai效率。rnai递送系统的选择可取决于sirna性质、靶细胞类型以及体内应用的递送途径。
在一些实施方案中,通过阳离子细胞穿透肽(cpp)递送rnai构建物。cpp已被用于包括蛋白质(例如,抗体)、肽、反义寡核苷酸和质粒dna的大分子的细胞内递送。除了利用传统的内吞途径之外,cpp-介导的rnai递送系统通过静电相互作用形成了非-共价复合物(非-共价cpp–sirna)或通过二硫键形成共价交联(共价cpp–sirna),并且可通过跨越细胞膜直接进入细胞。
在一些实施方案中,通过聚合物和树枝状载体递送rnai构建物。线性或分枝的阳离子聚合物是广为人知的dna和rna的有效转染剂。这些带正电荷的聚合物通过如下方式工作:通过静电相互作用与带负电荷的核酸的磷酸盐形成复聚物(polyplex)。其它合适的sirna的聚合物载体包括胶束、纳米复合物、纳米胶囊和纳米凝胶。复聚物的性质(例如,尺寸、表面电荷和结构)取决于阳离子聚合物的正电荷与sirna的磷酸盐基团数量之比。诸如聚-l-赖氨酸、聚乙烯亚胺(pei)、聚-d,l-丙交酯-共-乙交酯(plga)、聚(氰基丙烯酸烷基酯)、壳聚糖和明胶的多种聚合物是适合的。其他包括带正电荷表面基团的树枝状大分子,其精确的核-壳纳米结构能够通过内部包封、表面吸附或化学缀合来加载药物。示例性的树枝状大分子包括阳离子聚酰胺基胺(pamam,氨基封端表面),其任选乙二醇化以改善树枝状大分子的表面特征;聚(丙烯亚胺)(ppi)和含环糊精的阳离子聚合物和树枝状大分子。
在一些实施方案中,rnai构建物作为生物缀合物递送。rnai构建物可与多种分子[包括小分子(例如,胆固醇、胆汁酸和脂质)、肽(诸如阳离子细胞-穿透肽)、聚合物(诸如内渗剂两亲性聚(乙烯醚)pbave)、蛋白质(例如,抗体)和适体(例如,rna适体)]缀合以改善稳定性、细胞内化或细胞特异性活性靶向递送。
在一些实施方案中,rnai构建物与脂质-基载体一起递送,包括脂质体、胶束、微乳液和固体脂质纳米颗粒。脂质体因其相对简单和公知的药学特性是广为使用的sirna载体。癌症药物的若干脂质体载体已在人身上显示了良好的安全记录,已有公司(doxil)收到了人类用途的fda批准。脂质-基载体也已成功用于递送sirna(例如,在阳离子和促融合脂质的脂质体中负载的sirna的静脉或腹膜内注射)到内皮、res器官(例如,肝脏)和实体肿瘤中的靶位点。
在一些实施方案中,使用分子特洛伊木马(诸如特洛伊木马脂质体(thl)制剂)和亲和素-生物素技术递送rnai构建物。分子特洛伊木马也可配制为特洛伊木马脂质体以在体内递送表达shrna的质粒dna到脑。类似于非-病毒基因治疗的递送,编码短发夹rna(shrna)的质粒dna也可通过用聚乙二醇化的免疫脂质体(pil)进行静脉施用而被递送到脑。例如,质粒dna可被包封在脂质体(诸如100nm脂质体)中,所述脂质体可被聚乙二醇化并与受体特异性靶向单克隆抗体(mab)缀合。使用这种递送方式,用pil每周静脉施用rnai能够对人表皮生长因子(egfr)进行90%敲减,这导致患有颅内脑癌的小鼠的存活时间增加90%。也可使用相同技术递送包括sirna和mirna的rnai构建物至脑。例如,sirna可平行于靶向mab和链霉亲和素的缀合物的生产进行单生物素化。
分子特洛伊木马(mth)是经历了受体介导的跨血脑屏障(bbb)的输送(rmt)的内源性肽或模拟肽单克隆抗体(mab)。模拟肽mab结合bbb受体上的面外表位,这允许mab经历跨bbb的rmt而不会破坏内源性配体的bbb输送。模拟肽mab可通过bbbrmt系统携带任何附着药物甚至质粒dna跨越bbb。
已开发了一组物种特异性mab分子特洛伊木马用于脑药物递送。例如,对于小鼠中的药物递送,使用小鼠tfr的大鼠8d3mab。这种mth在大鼠中无活性。对于大鼠中的脑药物递送,使用大鼠tfr的鼠ox26mab,这种mth在小鼠或其他物种中无活性。对于向诸如恒河猴的旧大陆灵长类动物的脑药物递送,使用人胰岛素受体(hir)的鼠83-14mab。hirmab在诸如松鼠猴的新世界灵长类动物中无活性。已生产了hirmab的基因工程形式(包括嵌合hirmab和人化hirmab)以使得能在人中进行脑药物递送。
通过用分子特洛伊木马静脉施用向脑递送蛋白质治疗剂对于许多实验系统已进入了体内药理学实践。参见,pardridge(advdrugdelivrev.59(2-3):141-152,2007)。通过静脉施用在体内将重组蛋白递送到脑的这些研究证明了bbbmth可在体内将大分子治疗剂递送到脑。
对于非-病毒质粒dna的体内递送,开发了特洛伊木马脂质体(thl)制剂,其中mth以在体内稳定的方式与非-病毒质粒dna相关。具体地,将单个质粒dna分子包封在~100nm脂质体的内部,其表面缀有数千个聚合物链,诸如2000da聚乙二醇(peg)。1-2%的peg链的尖端缀有受体(r)-特异性mab,后者用作mth。这获得了包封质粒dna的聚乙二醇化免疫脂质体(pil)的制剂。靶向mab结合bbb受体以触发从血液到脑间质液的rmt。随后,靶向mab结合脑细胞的相同受体以触发进入脑细胞内空间的受体-介导的内吞作用。在胰岛素受体(其通常递送其内源性配体胰岛素到细胞核区域)的情况中,胰岛素受体递送质粒dna至脑细胞的细胞核,随后是内源性转基因的表达。在常规制剂中,有30-80个mab分子缀合到单个脂质体上。通过核酸酶处理可完全去除没有被完全包封在脂质体内部的任何dna。
使用这种技术,已通过如下方式将荧光素酶和β-半乳糖苷酶报告基因(以及类似形式的许多其他基因)用特洛伊木马脂质体递送到小鼠的脑中:以每只小鼠0.2ml体积中5μg质粒dna的量静脉注射到成年小鼠。通过静脉使用这种非病毒制剂,在整个脑中存在转基因的全局表达。表达的方式与在脑中普遍存在的神经元转铁蛋白受体的表达平行。转基因在皮质和皮质下结构中、在脉络丛中、在海马体、中脑、脊髓中表达,并且在小脑的浦肯野(purkinje)细胞层中高度表达。特洛伊木马脂质体技术使得能够在静脉施用非病毒制剂之后的24小时之内实现“成年转基因”。基因被递送到脑中的基本所有细胞。以10μg质粒dna/kg体重的剂量,可将约3-4个质粒dna分子递送到成年灵长类动物脑的每个脑细胞中。
在类似研究中,可将酪氨酸羟化酶(th)cdna配制在由sv40启动子驱动的表达质粒中。将th表达质粒包封在特洛伊木马脂质体中,所述特洛伊木马脂质体用大鼠tfr的小鼠ox26mab靶向大鼠的脑。用特洛伊木马脂质体进行th基因治疗导致病变的同侧纹状体th酶活性完全正常化,并且导致阿扑吗啡诱导的异常旋转行为减少82%。参见pardridge(advdrugdelivrev.59(2-3):141-152,2007)。
在一些实施方案中,使用组织特异性启动子驱动和限制转基因(例如,rnai构建物或aso)在脑中的表达。可用于本发明中的一种脑特异性启动子可具有人胶质原纤维酸性蛋白(gfap)基因的5’-侧翼序列(fs),以消除转基因在周围(非-cns)组织中的表达。因此,组织特异性启动子和特洛伊木马脂质体递送技术的组合使用允许治疗基因在体内的表达定位于特定器官或组织类型,诸如脑。
已使用iv注射rnai构建物(包封在用tfrmab靶向的特洛伊木马脂质体中)在脑中递送rnai构建物(shrna-编码质粒dna)的表达,并实现了脑肿瘤中靶基因表达的90%敲减。对侧脑中没有可检测的靶基因活性,并且rnai基因治疗对于非靶基因的表达没有作用。参见pardridge(advdrugdelivrev.59(2-3):141-152,2007)。因此,通过将rnai技术与特洛伊木马脂质体靶向技术相结合,可以实现rnai的体内治疗作用。
反义寡脱氧核苷酸(aso)、反义肽核酸(pna)或sirna的递送需要这些试剂与分子特洛伊木马的高亲和性连接。mth与核酸治疗剂之间的结合在体内在循环中必须是稳定的。在一些实施方案中,这样的核酸可经由聚阳离子桥(诸如聚赖氨酸或鱼精蛋白)连接到靶向配体。或者可选地,在其他实施方案中,这种核酸治疗剂可通过使用亲和素-生物素技术连接到靶向配体。
亲和素或链霉亲和素与生物素之间的结合非常牢固,并且不会被血清蛋白破坏。可在一个小瓶中配制靶向mab与亲和素或链霉亲和素(sa)的缀合物。在第二个小瓶中,制备单生物素化反义试剂(aso)或rnai/sirna。这两个小瓶混合之后立即用于静脉施用。由于生物素或sa与亲和素结合的极高亲和性,在反义试剂或sirna与靶向mab之间立即形成缀合物。生物素与亲和素或sa结合的解离半衰期为89日,解离常数是10-15m。因此,体内循环中反义试剂与靶向mab之间的结合在静脉施用之后数小时保持完整。反义试剂与靶向mab通过亲和素-生物素键的连接对反义试剂与靶rna的杂交没有抑制作用。在先前的研究中通过rna酶保护测定和northern印记证明了这一点。
使用这种技术,已使用mth将aso试剂(包括硫代磷酸酯(ps)和肽核酸(pna),诸如单生物素化pna)以及rnai构建物(诸如单生物素化sirna)递送到脑。
在一些实施方案中,使用sa与分子特洛伊木马的化学缀合物替代单生物素化sirna。在又另一个实施方案中,使用mab-亲和素融合蛋白。单生物素化sirna以及mth-sa或mth-亲和素缀合物可在静脉施用之前混合。mth可携带sirna分子跨越bbb和bcm,类似于前文中对于pna反义试剂所证明的那样。
在另一个实施方案中,降低或抑制plp1或促进plp1表达的plp1调控元件的表达的基因沉默剂可包括反义寡核苷酸(aso)。反义寡核苷酸是与编码特定蛋白质的mrna的编码链(有义链)互补(或反义)的相对短的核酸。尽管反义寡核苷酸通常为rna基的,但其也可以是dna基的。另外,通常对反义寡核苷酸进行修饰以提高其稳定性。
相信这些相对短的寡核苷酸与mrna的结合诱导了双链rna的延申,其触发内源性rna酶对信息的降解。另外,有时寡核苷酸被特别设计为在信息的启动子附近结合,在这些情况下,反义寡核苷酸可额外地干扰信息的翻译。无论反义寡核苷酸发挥作用的具体机制如何,将其施用于细胞或组织都允许编码特定蛋白质的mrna的降解。因此,反义寡核苷酸降低了特定蛋白质(例如,plp1)的表达和/或活性。
寡核苷酸可为dna或rna或其嵌合混合物或衍生物或修饰形式,可为单链或双链的。可在碱基部分、糖部分或磷酸盐主链对寡核苷酸进行修饰,例如,以改善分子稳定性、杂交等。寡核苷酸可包括其他附加基团,诸如肽(例如,为靶向宿主细胞受体),或者促进跨细胞膜(参见,例如,procnatlacadsci86:6553-6556;procnatlacadsci84:648-652;pct公开号wo88/09810,1988年12月15日公开)或跨血脑屏障(参见,例如,pct公开号wo89/10134,1988年4月25日公开)转运的试剂,杂交触发切割试剂(参见,例如,biotechniques6:958-976)或嵌入剂。(参见,例如,pharmres5:539-549)。为此,可将寡核苷酸与另一个分子缀合或偶联。
可通过本领域已知的标准技术合成本发明所述的寡核苷酸,例如,使用自动化dna合成器(诸如可商购自biosearch,appliedbiosystems等的)。例如,可通过stein等(nucl.acidsres.16:3209的方法合成硫代磷酸酯寡核苷酸,可通过使用受控孔玻璃聚合物载体制备甲基磷酸酯寡核苷酸(procnatlacadsci85:7448-7451)。
本领域技术人员可进行合适寡核苷酸的选择。如果核酸序列编码特定蛋白质,本领域技术人员可设计结合该蛋白质的反义寡核苷酸并在体外或体内系统测试这些寡核苷酸以确定其结合编码该特定蛋白质的mrna并介导其降解。为设计特异性结合特定蛋白质并介导其降解的反义寡核苷酸,重要的是寡核苷酸所识别的序列对于该特定蛋白质来说是独特或基本独特的。例如,经常在蛋白质上重复的序列不是设计特异性结合并降解特定信息的寡核苷酸的理想选择。本领域技术人员可设计寡核苷酸并比较该寡核苷酸的序列与公众可获得的数据库中所保藏的核酸序列以确定该序列对特定蛋白质是特异性或基本特异性的。
已开发了多种方法递送反义dna或rna至细胞;例如,可将反义分子直接注入组织位点,或者全身性施用被设计为靶向所需细胞的修饰反义分子(例如,与特异性结合靶细胞表面上表达的受体或抗原的肽或抗体反义连接)。
然而,在一些情况中难以实现足以抑制内源性mrna上的翻译的反义寡核苷酸的细胞内浓度。因此,另一种方法利用了重组dna构建物,其中反义寡核苷酸被置于强poliii或polii启动子的控制下。例如,可体内引入载体以使其被细胞摄入并指导反义rna的转录。这样的载体可保持附加型或变为染色体整合型,只要其可被转录以产生所需的反义rna。这样的载体可通过本领域中标准的重组dna技术构建。载体可为质粒、病毒或本领域中已知用于哺乳动物细胞中的复制和表达的其他品种。
编码反义rna的序列的表达可通过本领域中已知的启动子在哺乳动物、优选人的细胞中发挥作用。这样的启动子可为诱导型或组成型的。这样的启动子包括但不限于:sv40早期启动子区域(nature290:304-310),劳氏肉瘤病毒的3'长末端重复中所含的启动子(cell22:787-797),疱疹胸苷激酶启动子(procnatlacadsci78:1441-1445),金属硫蛋白基因的调控序列(nature296:39-42)等。可使用一种类型的质粒、粘粒、yac或病毒载体来制备可被直接引入到组织位点的重组dna构建物。或者可选地,可使用选择性感染所需组织的病毒载体,在这种情况中可通过另一种途径施用(例如,全身性地)。
细胞
因此,本发明提供了在plp1基因中包含插入或缺失(插入缺失)的基因修饰细胞,例如plp1中外显子1或3的核苷酸序列的靶向缺失或者失活或降低plp1表达的其他修饰。细胞优选产生功能性髓鞘或是产生髓鞘的细胞的祖细胞。在某个实施方案中,细胞是受精卵、卵子、神经元干细胞(nsc)、opc或少突胶质细胞。在一些实施方案中,基因修饰细胞包含在plp2的外显子3的5'末端的约80个核苷酸的靶向缺失,使plp1表达失活。还提供了其中对涉及plp1转录或翻译的plp1调控元件进行修饰的基因修饰细胞。
通常使用一种或多种核酸酶将插入缺失(插入或缺失)以靶向方式整合到细胞的基因组中。在一些实施方案中,插入缺失被整合到plp1中,例如用于使plp1基因失活。在一些实施方案中,在通过核酸酶引入双链断裂(dsb)(例如,在plp1基因或其调控元件中)之后,由于nhej或其他修复机制产生插入缺失。在其他实施方案中,将插入缺失整合到与plp1相关的内源性基因组基因座中,例如plp1基因调控元件,如增强子或启动子。在本发明所述的任何细胞中,可整合入外显子和/或内含子(例如,plp1的外显子1或3)中。
与随机整合和缺失不同,靶向整合或缺失确保了插入缺失被整合到指定基因。可将插入缺失整合到靶基因的任何地方。在一些实施方案中,将插入缺失或供体序列整合到核酸酶切割位点或其附近,例如,在切割位点上游或下游的1-3000个碱基对(或其间的任何值)内,更优选在切割位点任一侧的1-1000个碱基对(或其间的任何值)内或切割位点任一侧的1-500个碱基对(或其间的任何值)内或1-100个碱基对(或其间的任何值)内。在一些实施方案中,包含供体转基因的整合序列不包含任何载体序列(例如,病毒载体序列)。
任何细胞类型可如本发明所述地进行基因修饰以包含失活或降低plp1表达的序列,包括但不限于细胞和细胞系。本发明所述细胞的其他非限制性实例包括受精卵、神经干细胞(nsc)、少突胶质细胞祖细胞(opc)、神经元细胞和胶质细胞诸如少突胶质细胞、星形胶质细胞、室管膜细胞或小神经胶质细胞。本发明所述细胞的其他非限制性实例包括自体同源(例如,来源于患者的)或异源性(同种异体)的多能、全能或专能干细胞(例如,胚胎干细胞等)。在一些实施方案中,本发明所述细胞为opc。
本发明所述细胞可用于治疗和/或预防患有障碍的受试者的髓鞘相关障碍,例如,通过离体治疗。可使用标准技术扩增核酸酶-修饰的细胞,随后将其再引入患者体内。参见,例如,tebas等(2014)newengjmed370(10):901。在干细胞的情况中,在融合入患者体内之后,还发生了这些前体向表达失活plp1的细胞中的体内分化。提供了包含本发明所述细胞的药物组合物。另外,在施用给患者之前可将细胞低温贮藏。
本发明所述细胞和离体方法提供了对受试者的髓鞘相关障碍的治疗和/或预防,并且消除了对连续施用预防性药物或有风险的疗法的需要。因此,本发明提供了更安全、成本有效和时间有效的治疗和/或预防髓鞘相关障碍的方法。
递送
可通过任何合适的方式递送本发明所述的核酸酶、编码这些核酸酶的多核苷酸、供体多核苷酸和包含蛋白质、寡核苷酸和/或多核苷酸的组合物。在一些实施方案中,在体内递送核酸酶和/或供体。在其他实施方案中,将核酸酶和/或供体递送到分离的细胞(例如,自体同源或异源干细胞)中以提供可用于体外递送到髓鞘相关障碍患者的修饰细胞。
本发明所述的递送核酸酶的方法如例如以下文献中所述:美国专利6,453,242;6,503,717;6,534,261;6,599,692;6,607,882;6,689,558;6,824,978;6,933,113;6,979,539;7,013,219;和7,163,824,上述所有文献的公开内容通过引用以其整体并入本发明。
也可使用任何核酸递送机制来递送本发明所述的核酸酶和/或供体构建物,包括裸dna和/或rna(例如,mrna)以及包含编码一个或多个组成部分的序列的载体。可使用任何载体系统,包括但不限于质粒载体、dna微环、逆转录病毒载体、慢病毒载体、腺病毒载体、痘病毒载体;疱疹病毒载体和腺相关病毒载体等及其组合。还参见美国专利6,534,261;6,607,882;6,824,978;6,933,113;6,979,539;7,013,219;和7,163,824,以及美国专利申请序列号14/271,008,上述文献通过引用以其整体并入本发明。此外,很明显任何这些系统可包含治疗所需的一个或多个序列。因此,当将一种或多种核酸酶和供体构建物引入细胞中时,可将核酸酶和/或供体多核苷酸携带在相同递送系统或不同递送机制上。当使用多种系统时,每个递送机制可包含编码一个或多个核酸酶或/或供体构建物的序列(例如,编码一种或多种核酸酶的mrna和/或携带一个或多个供体构建物的mrna或aav)。
合适的载体可包括非整合、非免疫原性并且能够感染分裂细胞和休眠细胞的递送载体。在一些实施方案中,cas9核酸酶和sgrna的体内递送可由腺相关病毒(aav)载体介导。用于体内递送的aav载体包括具有穿透受试者的中枢神经系统和感染中枢神经系统的产生胶质髓鞘的细胞类型(少突胶质细胞、opc、npc等)的能力的aav血清型。在一个示例性实施方案中,采用aav载体进行cas9核酸酶(例如,sacas9或spcas9核酸酶)的体内递送。在一些实施方案中,将核酸酶cas9和sgrna包装到一个aav载体中用于细胞中plp1的核酸酶-介导的基因破坏。详情如下。
可使用常规的基于病毒和非-病毒的基因转移方法将编码核酸酶和供体构建物的核酸引入细胞(例如,哺乳动物细胞)和靶组织中。非-病毒载体递送系统包括dna质粒、dna微环、裸核酸和与诸如脂质体、脂质纳米颗粒(lnp)、聚乳酸-乙醇酸纳米颗粒、聚胺复合剂或泊洛沙姆的递送载体复合的核酸。病毒载体递送系统包括dna和rna病毒,其在递送到细胞之后具有附加型或整合型基因组。对于基因治疗过程的综述,参见以下:anderson,science256:808-813(1992);nabel&felgner,tibtech11:211-217(1993);mitani&caskey,tibtech11:162-166(1993);dillon,tibtech11:167-175(1993);miller,nature357:455-460(1992);vanbrunt,biotechnology6(10):1149-1154(1988);vigne,restorativeneurologyandneuroscience8:35-36(1995);kremer&perricaudet,britishmedicalbulletin51(1):31-44(1995);haddada等,currenttopicsinmicrobiologyandimmunologydoerfler和
核酸的非病毒递送方法包括电穿孔、脂质转染、显微注射、基因枪、病毒体、脂质体、免疫脂质体、聚阳离子或脂质:核酸缀合物、裸dna、裸rna、封端rna、人工病毒体和试剂增强的dna摄取。使用例如sonitron2000系统(rich-mar)的声穿孔方法也可用于递送核酸。
其他示例性的核酸递送系统包括以下中提供的那些:amaxabiosystems(cologne,germany),maxcyte,inc.(rockville,md.),btxmoleculardeliverysystems(holliston,mass.)和copernicustherapeuticsinc(参见例如美国专利6,008,336)。脂质转染如例如美国专利5,049,386;4,946,787;和4,897,355中所述,脂质转染试剂有商业售卖(例如,transfectamtm和lipofectintm)。适合用于多核苷酸的有效的受体识别脂质转染的阳离子和中性脂质包括felgner、wo91/17424、wo91/16024中的那些。在一些方面,核酸酶作为mrna递送(例如,使用电穿孔),转基因经由诸如病毒载体、微环dna、质粒dna、单链dna、线性dna、脂质体、纳米颗粒等的其他形式递送。
包含靶向脂质体(诸如免疫脂质复合物)的脂质:核酸复合物的制备是本领域技术人员公知的(参见,例如,crystal,science270:404-410(1995);blaese等,cancergenether.2:291-297(1995);behr等,bioconjugatechem.5:382-389(1994);remy等,bioconjugatechem.5:647-654(1994);gao等,genetherapy2:710-722(1995);ahmad等,cancerres.52:4817-4820(1992);美国专利4,186,183,4,217,344,4,235,871,4,261,975,4,485,054,4,501,728,4,774,085,4,837,028,和4,946,787)。
其他递送方法包括使用将要被递送的核酸包装到engeneic递送载体(edv)中。使用双特异性抗体将这些edv特异性递送到靶组织,其中所述抗体的一个臂具有对靶组织的特异性,且另一个具有对edv的特异性。抗体将edv带到靶细胞表面,随后通过内吞作用将edv带入细胞中。一旦进入细胞,则释放内容物(参见macdiarmid等(2009)naturebiotechnology27(7):643)。
使用基于rna或dna病毒的系统递送编码工程化crispr/cas系统或编码可在靶细胞中产生sirna、shrna或mirna的rnai构建物的核酸利用了高度进化的过程以将病毒靶向体内的特定细胞并将病毒负载运输到细胞核。可将病毒载体直接施用给受试者(体内),或者其可用于在体外治疗细胞并且将修饰细胞施用给受试者(离体)。用于递送crispr/cas系统的常规的基于病毒的系统包括但不限于用于基因转移的逆转录病毒,慢病毒,腺病毒,腺相关、牛痘和单纯疱疹病毒载体。使用逆转录病毒、慢病毒和腺相关病毒基因转移方法可进行宿主基因组中的整合,通常导致插入的转基因的长期表达。另外,已经在许多不同细胞类型和靶组织中观察到高转导效率。
可通过插入外来包膜蛋白改变逆转录病毒的趋向性,扩增靶细胞的潜在靶群。慢病毒载体是能够转导或感染非分裂细胞并且通常产生高病毒滴度的逆转录病毒载体。逆转录病毒基因转移系统的选择取决于靶组织。逆转录病毒载体由顺式作用的长末端重复组成,具有高达6-10kb外来序列的包装能力。最小顺式作用ltr足以用于载体的复制和包装,然后将其用于将治疗基因整合到靶细胞中以提供永久性转基因表达。广泛使用的逆转录病毒载体包括基于以下的那些:鼠白血病病毒(mulv),长臂猿白血病病毒(galv)、猿猴免疫缺陷病毒(siv)、人免疫缺陷病毒(hiv)及其组合(参见,例如,buchscher等,j.virol.66:2731-2739(1992);johann等,j.virol.66:1635-1640(1992);sommerfelt等,virol.176:58-59(1990);wilson等,j.virol.63:2374-2378(1989);miller等,j.virol.65:2220-2224(1991);pct/us94/05700)。
腺病毒类载体在许多细胞类型中能够有极高的转导效率,并且无需细胞分裂。使用这种载体,已获得了高滴度和高表达水平。这种载体可在相对简单的系统中大量生产。腺相关病毒(“aav”)载体也用于用靶核酸转导细胞,例如,用于核酸和肽的体外生产以及体内和离体基因治疗程序(参见,例如,west等,virology160:38-47(1987);美国专利4,797,368;wo93/24641;kotin,humangenetherapy5:793-801(1994);muzyczka,j.clin.invest.94:1351(1994))。重组aav载体的构建在许多公开出版物中有所描述:包括:美国专利5,173,414;tratschin等,mol.cell.biol.5:3251-3260(1985);tratschin,等,mol.cell.biol.4:2072-2081(1984);hermonat&muzyczka,pnas81:6466-6470(1984);和samulski等,j.virol.63:03822-3828(1989)。可使用任何aav血清型(包括aav1、aav3、aav4、aav5、aav6和aav8、aav8.2、aav9和aavrh10)以及假aav(诸如aav2/8、aav2/5和aav2/6)。其他腺病毒类载体包括在静脉注射之后有效和广泛转导哺乳动物中枢神经系统(cns)的aav变体,诸如在deverman等(2016)naturebiotechnology204-209中描述的aav-php.b载体。
目前临床试验中至少有六种病毒载体方法可用于基因转移,其利用涉及通过插入辅助细胞系的基因互补缺陷载体以产生转导剂的方法。
plasn和mfg-s是已用于临床试验中的逆转录病毒载体的实例(dunbar等,blood85:3048-305(1995);kohn等,nat.med.1:1017-102(1995);malech等,pnas94:2212133-12138(1997))。pa317/plasn是在基因治疗试验中使用的第一种治疗载体。(blaese等,science270:475-480(1995))。对于mfg-s包装的载体已观察到50%或更高的转导效率。(ellem等,immunolimmunother.44(1):10-20(1997);dranoff等,hum.genether.1:111-2(1997)。
重组腺相关病毒载体(raav)是基于有缺陷和非致病性细小病毒腺相关病毒2型病毒的有前景的替代性基因递送系统。所有载体均衍生自仅保留转基因表达盒侧翼的aav145碱基对(bp)末端反向重复的质粒。由于整合到转导细胞的基因组中,有效的基因转移和稳定的转基因递送是这种载体系统的关键特征。(wagner等,lancet351:91171702-3(1998),kearns等,genether.9:748-55(1996))。根据本发明也可使用其他aav血清型(包括aav1、aav3、aav4、aav5、aav6、aav8、aav9和aavrh10)及其所有变体(包括从文库选择的工程化突变体)。
复制缺陷型重组腺病毒载体(ad)也可以高滴度制备并且容易感染多种不同的细胞类型。大部分腺病毒载体被工程化以使得转基因替代ade1a、e1b、和/或e3基因;随后,复制缺陷型载体在人293细胞中繁殖,所述细胞以反式(intrans)提供缺失的基因功能。ad载体能在体内转导多种类型的组织,包括非分裂的分化细胞,诸如在肝脏、肾脏和肌肉中发现的那些。常规的ad载体具有巨大的携带能力。在临床试验中使用ad载体的实例涉及用肌肉内注射进行抗肿瘤免疫的多核苷酸治疗(sterman等,hum.genether.7:1083-9(1998))。在临床试验中使用腺病毒载体进行基因转移的其他实例包括:rosenecker等,infection24:15-10(1996);sterman等,hum.genether.9:71083-1089(1998);welsh等,hum.genether.2:205-18(1995);alvarez等,hum.genether.5:597-613(1997);topf等,genether.5:507-513(1998);sterman等,hum.genether.7:1083-1089(1998)。
使用包装细胞形成能够感染宿主细胞的病毒颗粒。这样的细胞包括293细胞(其包装了腺病毒)和ψ2细胞或pa317细胞(其包装了逆转录病毒)。用于基因治疗的病毒载体通常通过将核酸载体包装到病毒颗粒中的生产细胞系产生。载体通常包含包封和随后整合到宿主(如果适用的话)所需的最小病毒序列,其他病毒序列被编码要被表达的蛋白质的表达盒所替代。通过包装细胞系以反式供应缺失的病毒功能。例如,在基因治疗中使用的aav载体通常仅具有来自包装和整合到宿主基因组中所需的aav基因组的末端反向重复(itr)序列。病毒dna被包装到包含编码其他aav基因(即rep和cap)但缺少itr序列的辅助质粒的细胞系中。细胞系也用作为助手的腺病毒感染。辅助病毒促进了aav载体的复制和来自辅助质粒的aav基因的表达。由于缺乏itr序列,没有大量包装辅助质粒。可通过例如腺病毒比aav更敏感的热处理来减少腺病毒的污染。
在许多基因治疗应用中,理想的是以高特异性程度将基因治疗载体递送到特定组织类型。因此,通过在病毒外表面上表达作为与病毒外壳蛋白的融合蛋白的配体来修饰病毒载体以使其对指定细胞类型具有特异性。选择配体使其对已知存在于所关注的细胞类型上的受体具有亲和性。例如,han等,proc.natl.acad.sci.usa92:9747-9751(1995)报告了莫洛尼鼠白血病病毒可被修饰以表达与gp70融合的人调节蛋白,并且该重组病毒感染一些表达人表皮生长因子受体的人乳腺癌细胞。该原理可以扩展到其他病毒-靶细胞对,其中靶细胞表达受体,病毒表达包含细胞表面受体配体的融合蛋白。例如,可以将丝状噬菌体工程化以展示对几乎任何所选择的细胞受体具有特异性结合亲和性的抗体片段(例如,fab或fv)。尽管以上描述主要适用于病毒载体,但相同的原理可应用于非病毒载体。可以将这些载体工程化为含有特异性摄取序列,其有利于特定靶细胞的摄取。
可通过对单个受试者施用来体内递送基因治疗载体,通常通过全身施用(例如,静脉内、腹膜内、肌肉内、皮下、舌下或颅内输注)、如下所述的局部应用或经由肺部吸入施用。或者可选地,可将载体离体递送到细胞中,诸如从单个患者外植的细胞(例如,opc)或通用型供体神经干细胞,随后将细胞再植入患者体内,通常在选择包含载体的细胞之后。
包含核酸酶和/或供体构建物的载体(例如,逆转录病毒、腺病毒、aav、脂质体等)也可被直接施用给生物体以体内转导细胞。或者可选地,可施用裸dna。通过通常用于将分子引入与胶质细胞最终接触的任何途径来施用,包括但不限于注射、输注、局部应用、吸入和电穿孔。施用这种核酸的合适方法是本领域技术人员可获得和公知的,尽管可使用多于一种途径施用特定组合物,但特定途径经常可提供比另一途径更立即和更有效的反应。
适合引入本发明所述的多核苷酸的载体包括非整合的慢病毒载体(idlv)。参见ory等(1996)proc.natl.acad.sci.usa93:11382-11388;dull等(1998)j.virol.72:8463-8471;zuffery等(1998)j.viro.72:9873-9880;follenzi等(2000)naturegenetics25:217-222;美国专利公开2009/054985。
药学可接受的载体部分通过要被施用的特定组合物以及通过用于施用该组合物的特定方法确定。因此,存在多种可用的合适药物组合物制剂,如下所述(参见,例如,remington’spharmaceuticalsciences,17thed.,1989)。
显而易见的是可使用相同或不同的系统递送核酸酶-编码序列和供体构建物。例如,可通过aav携带供体多核苷酸,同时可通过mrna携带一种或多种核酸酶。此外,可通过相同或不同途径施用不同系统(肌肉内注射、尾静脉注射、其他静脉注射、腹膜内施用和/或肌肉内注射)。可同时或以任何依次顺序递送多种载体。
用于离体和体内施用的制剂包括在液体或乳化液体中的悬浮剂。活性成分通常与药学可接受的并且与活性成分可相容的赋形剂混合。合适的赋形剂包括,例如,水、盐水、葡萄糖、甘油、乙醇等及其组合。另外,组合物可包含少量辅助物质,诸如湿润剂或乳化剂、ph缓冲剂、稳定剂或其他增强药物组合物效率的试剂。
通过aav递送的crispr/cas介导的基因沉默
在一些实施方案中,本发明所述的方法可通过递送crispr/cas系统来实现,所述crispr/cas系统包括特异性靶向plp1基因或其调控元件或其部分的向导rna(sgrna)。
在一个实施方案中,该系统可采用2个aav载体以及任选的第三aav载体:一个编码cas9或其功能性直系同源物,一个包含用于靶向切割plp1基因或其调控元件的向导rna序列,以及任选地另一个包含要被插入到切割位点以修复或替代缺陷的plp1基因的突变plp1基因(诸如plp1点突变)的供体cdna序列。供体与cas9构建物的施用比可为1:1至5:1范围内的任意数值。
在另一个实施方案中,该系统可采用2个aav载体:一个编码长度小于3.5kb的cas9直系同源物并且将具有以顺式编码的向导rna,并且一个载体包含要在切割位点处插入的突变plp1基因的供体cdna序列。对于大于4.8kb的靶标,该供体可包含允许在大多数突变基因上游纠正的高达4.8kb的基因的3’cdna部分,或者5’启动子和基因的上游cdna部分,其随后被剪接到正确的下游序列。
在进一步的实施方案中,该系统采用2个aav载体:一个编码cas9或功能性直系同源物,并且一个包含特异性切割靶向基因的向导rna序列。
在又一个实施方案中,该系统将采用1个aav载体,所述载体包含编码功能性ii型crispr-cas9的核酸和特异性用于切割靶基因(例如,plp1)的向导rna。
在一个实施方案中,所述方法包括提供包含crispr系统的元件的一个或多个aav载体(通常为1、2或3个aav载体),以结合靶基因以实现对所述靶基因/多核苷酸的切割,从而修饰靶基因,诸如破坏靶基因或用供体核酸纠正或替换靶基因的全部或一部分。所述crispr系统的一部分包括crispr酶,其可与向导rna序列复合,所述向导rna可与所述靶基因中的靶序列杂交。
靶基因处的切割可包括通过crispr酶切割一条或两条链。在一些实施方案中,方法包括通过引入供体核酸来纠正或替代所切割的靶基因,所述供体核酸编码纠正突变或缺陷靶基因的蛋白质。
本发明所述的cas基因包括但不限于cas3或cas9。酶可为cas9同系物或直系同源物。cas9直系同源物可包括来自以下的cas9:化脓性链球菌(streptococcuspyogenes)、脑膜炎双球菌(neisseriameningitidis)、嗜热链球菌(streptococcusthermophilus)、肺炎链球菌(streptococcuspneumnoniae)、结肠弯曲菌(campylobactercoli)、空肠弯曲菌(campylobacterjejuni)、变异链球菌(streptococcusmutans)、多杀性巴氏杆菌(pasteurellamultocida)、长双歧杆菌(bifidobacteriumlongum)、史氏芽孢杆菌(bacillussmithii)、齿密螺旋体(treponemadenticola)、犬支原体(mycoplasmacanis)和粪肠球菌(enterococcusfaecalis)。cas9可包括源自这些生物体的突变cas9。
示例性的aav载体包括以下任一种的衣壳序列:aav1、aav2、aav3、aav4、aav5、aav6、aav7、aav8、aav9、aav10、aav11、rhlo、rh74或aav-2i8;或以下的衣壳变体:aav1、aav2、aav3、aav4、aav5、aav6、aav7、aav8、aav9、aav10、aav11、rhlo、rh74或aav-2i8。本发明的重组aav载体还包括aav1、aav2、aav3、aav4、aav5、aav6、aav7、aav8、aav9、aav10、aav11、rhlo、rh74或aav-2i8及其变体。
具体的衣壳变体包括以下的衣壳变体:aav1、aav2、aav3、aav4、aav5、aav6、aav7、aav8、aav9、aav10、aav11、rhlo、rh74或aav-2i8,诸如具有氨基酸取代、缺失或插入/添加的衣壳序列。
aav载体可包括以顺式或反式作用的其他元件。在特定实施方案中,包含载体基因组的aav载体还具有:位于供体序列的5’或3’末端侧翼的一个或多个末端反向重复(itr)序列;驱动供体序列的转录的表达控制元件(例如,启动子或增强子),诸如组成型或可调控控制元件或组织特异性表达控制元件;内含子序列、填充物(staffer)或填充多核苷酸序列;和/或位于供体序列3’的聚-腺嘌呤序列。
通常,表达控制元件是影响可操作连接的多核苷酸的表达的核酸序列。存在于载体内的控制元件(包括如本发明所述的表达控制元件,诸如启动子和增强子)被包含以促进适当的核酸转录和翻译(例如,启动子,增强子,内含子剪接信号,维持正确的基因阅读框以允许mrna的框内翻译,以及终止密码子等)和aav包装。这样的元件通常以顺式作用,称为“顺式作用”元件,但也可以反式作用。
可在转录、翻译、剪接、信息稳定性等水平上实现表达控制。调节转录的表达控制元件被并置在转录核酸的5’末端(即,“上游”)附件。表达控制元件也可位于转录序列的3’末端(即,“下游”)或转录物内(例如,在内含子中)。表达控制元件可定位成邻近或远离转录序列。通常,由于一些载体(诸如aav载体)的多核苷酸长度限制,这样的表达控制元件将距离转录核酸1-1000个核苷酸内。
本发明所用的“启动子”可操作地连接到相邻序列,并且与不存在启动子时表达的量相比,增加了由核酸表达的量。
本发明所用的“增强子”位于核酸附近,通常位于启动子元件上游但也起作用,并且可位于dna序列(例如,供体核酸)的下游或内部。增强子元件位于核酸上游或下游的100个碱基对、200个碱基对或者300或更多个碱基对。增强子元件通常还提高核酸的表达。
表达控制元件包括普遍存在的或混杂的启动子/增强子,其能够在许多不同细胞类型中驱动多核苷酸的表达。这样的元件包括但不限于巨细胞病毒(cmv)立即早期启动子/增强子序列,劳氏肉瘤病毒(rsv)启动子/增强子序列,以及在多种哺乳动物细胞类型中有活性的其他病毒启动子/增强子,或自然中不存在的合成元件(参见,例如,boshart等,cell,41:521-530(1985)),sv40启动子,二氢叶酸还原酶(dhfr)启动子,细胞质β-肌动蛋白启动子和磷酸甘油激酶(pgk)启动子。
表达控制元件还可包括在特定组织或细胞类型中有活性的那些,其在本发明中被称为“组织特异性表达控制元件/启动子”。组织特异性表达控制元件通常在特定细胞或组织(例如,眼睛、视网膜、中枢神经系统、脊髓、眼睛、视网膜等)中有活性。在一些实施方案中,组织特异性表达控制元件在cns中有活性,诸如神经元干细胞、opc、少突胶质细胞等。在一些实施方案中,组织特异性表达控制元件在受精卵或卵子中有活性。
表达控制元件通常在这些细胞、组织或器官中有活性,这是因为它们被转录激活蛋白或转录的其他调节因子识别,这对特定细胞、组织或器官类型是独特的。
表达控制元件也可以可调控方式赋予表达,即,信号或刺激增加或降低了可操作连接的核酸的表达。响应于信号或刺激而增加可操作连接的核酸的表达的可调控元件也被称为“可诱导元件”(即,通过信号诱导)。具体实例包括但不限于激素(例如,类固醇)诱导的启动子。响应于信号或刺激而降低可操作连接的核酸的表达的可调控元件被称为“可抑制元件”(即,信号降低了表达,因此当移除或不存在信号时,表达会增加)。通常,这样的元件所赋予的增加或降低的量与存在的信号或刺激的量成正比;信号或刺激的量越大,表达增加或降低得越多。
表达控制元件还包括天然元件。当希望核酸的表达可模拟天然表达时,可使用天然控制元件(例如,启动子)。当想要暂时性调控或发育性调控或以组织特异性方式调控或响应于特定的转录刺激来调控核酸表达时,可使用天然元件。也可使用其它天然表达控制元件,诸如内含子、多腺苷酸化位点或科扎克共有序列。
aav载体还可包括填充或填充物多核苷酸序列。例如,当供体核酸的长度小于约4.7kb时,填充或填充物多核苷酸序列的长度在与供体核酸组合时,总组合长度为约3.0-5.5kb或约4.0-5.0kb或约4.3-4.8kb。
填充或填充物多核苷酸序列可位于载体序列中任何所需位置,使其不阻止载体的功能或活性。在一个方面中,填充或填充物多核苷酸序列位于5’和/或3’itr(例如,aav1、aav2、aav3、aav4、aav5、aav6、aav7、aav8、aav9、aav10、aav11、rhlo、rh74或aav-2i8及其变体的itr)之间,所述itr位于供体核酸序列各自的5’和/或3’末端侧翼。
通常,填充或填充物多核苷酸序列是惰性或无害的,并且没有功能或活性。在多个特定方面,填充或填充物多核苷酸序列不是细菌多核苷酸序列,填充或填充物多核苷酸序列不是编码蛋白质或肽的序列,填充或填充物多核苷酸序列与以下任何的序列都不同:供体序列、aav末端反向重复(itr)序列、表达控制元件或多腺苷酸化(poly-a)信号序列。在多个特定方面中,填充或填充物多核苷酸序列是与供体序列相关或不相关的内含子序列。
内含子也可用作填充或填充物多核苷酸序列以实现包装到病毒颗粒中的aav载体的长度。用作填充或填充物多核苷酸序列的内含子和内含子片段(例如,fix的内含子1的部分)也可增强表达。与不存在内含子元件时的表达相比,包含内含子元件可增强表达(kurachi等,1995,出处同上)。
内含子的使用不限于天然基因组序列,并且可包括与完全不同的基因或其它dna序列相关的内含子。因此,核酸的其他未翻译(非-蛋白质编码)区域,诸如在来自同源(相关)基因和非同源(不相关)基因的基因组序列中发现的内含子,也可用作根据本发明的填充或填充物多核苷酸序列。
供体核酸、表达控制元件、itr、polya序列、填充或填充物多核苷酸序列的长度可以不同。在特定方面中,序列的长度为约1-10、10-20、20-30、30-40、40-50、50-60、60-75、75-100、100-150、150-200、200-250、250-300、300-400、400-500、500-750、750-1,000、1,000-1,500、1,500-2,000、2,000-2,500、2,500-3,000、3,000-3,500、3,500-4,000、4,000-4,500、4,500-5,000或更多个核苷酸,其长度高达aac包装尺寸限制的极限。
crispr/cas系统包括cas9编码序列,并且可通过aav载体将sgrna引入/转移/转导/转染到靶细胞中。术语“转导”和“转染”是指将诸如核酸的分子引入到细胞或宿主生物体中。因此,转导细胞(例如,在哺乳动物中,诸如细胞或组织或器官细胞)意味着在将外源分子(例如,多核苷酸或蛋白质(例如,转基因))引入细胞中之后细胞中的基因变化。因此,“转导”细胞是已将外源分子引入其中或其子代中的细胞。在本发明的方法和用途中,转导细胞可在受试者中,例如,体内或离体。
本发明的方法和用途提供了将crispr/cas/sgrna以及任选的供体核酸(转基因)递送(转导)到宿主细胞(包括分裂和/或非分裂细胞)的方式。本发明的aav载体、方法、用途和药物制剂还可用于将核酸或蛋白质递送、施用或提供给有需要的受试者作为治疗方法的方法。以此方式,转录核酸并在受试者体内产生蛋白质。受试者可受益于该核酸或蛋白质或需要该核酸或蛋白质,因为受试者缺乏该核酸或蛋白质,或者因为在受试者中生产该核酸或蛋白质会作为治疗方法或其它而带来一些治疗效果。
在多个实施方案中,将aav载体递送到受试者的真核细胞中。受试者通常为动物,并且包括人和兽医学用途。因此合适的受试者包括哺乳动物,诸如人和非人哺乳动物(例如,灵长类动物)。其它受试者包括灵长类动物(猿猴、长臂猿、大猩猩、黑猩猩、猩猩、猕猴)、家养动物(狗和猫)、农场动物(禽类,诸如鸡和鸭,马,牛,山羊,绵羊,猪)和实验动物(小鼠、大鼠、兔、豚鼠)。人类受试者包括胎儿、新生儿、婴儿、青少年和成年受试者。例如,受试者可以是低于20岁、15岁、10岁、5岁、3岁、2岁、1岁、6个月、3个月或1个月的人。受试者可还包括动物疾病模型,例如,血液凝固疾病的小鼠和其它动物模型以及本领域技术人员已知的其它模型。
适合治疗的受试者包括具有如下那些:其具有功能性基因产物(蛋白质)产量不足或处于此风险中,或者具有功能性基因产物(蛋白质)缺乏,或者产生异常的、部分功能性的或无功能的基因产物(蛋白质),这可导致疾病。在特定实施方案中,将受益于或需要破坏、纠正或替代缺陷基因(例如,plp1)或者需要破坏、纠正或替代编码具有缺陷或只具有部分功能或活性的蛋白质的基因的受试者。
因此,治疗的治疗效果或有益效果是为特定受试者提供的客观或主观的可测量或可检测的改善或利益。治疗效果或有益效果可以但不必是完全消除疾病的全部或任何特定的不良症状、障碍、患病或并发症。因此,当在短或长时间(小时、日、周、月等)内由疾病引起或与其相关的不良症状、障碍、患病或并发症逐渐改善或部分减少,或者由疾病引起或与其相关的一种或多种不良症状、障碍、患病或适应症的恶化或进展被抑制、降低、减少、压制、预防、限制或者控制,则实现了令人满意的临床终点。
实现治疗效果的剂量,例如,以载体基因组/千克体重(vg/kg)计的剂量,可根据若干因素而变,包括但不限于:施用途径、实现治疗效果所需的核酸表达、所治疗的具体疾病、对载体的任何宿主免疫反应以及表达蛋白质的稳定性。本领域技术人员可根据上述因素以及其它因素来确定治疗患有特定疾病或障碍的患者的raav/载体基因组剂量范围。
可在发展出因疾病引起或与其相关的不良症状、病症、适应症等之前实施对受试者的体内施用或递送。例如,可使用筛选(例如,基因筛选)来鉴定作为本发明组合物、方法和用途的候选人的这种受试者。因此,这样的受试者包括被筛选出来对功能性基因产物(蛋白质)量的不足或缺乏呈阳性或者产生异常的、部分功能性或无功能性基因产物(蛋白质)的那些。
施用或递送的方法包括与受试者相容的任何方式。本发明的方法和用途包括全身性、区域性或局部或通过任何途径(例如,通过注射或输注)递送和施用。这样的递送和施用包括肠胃外,例如,眼内、血管内、静脉内、肌肉内、腹膜内、皮内、皮下或经粘膜。示例性的施用和递送途径包括静脉内(i.v.)、腹膜内(i.p.)、动脉内、皮下、胸膜内、插管、肺内、腔内、离子电渗疗法、器官内、淋巴管内。在特定实施方案中,aav载体经肠胃外施用或递送,诸如静脉内、动脉内、眼内、肌肉内、皮下或通过导管或插管。
剂量可以变化并且取决于治疗所涉及的疾病的类型、发作、进展、严重程度、频率、持续时间或概率,所需临床终点,先前或同时的治疗,受试者的一般健康情况、年龄、性别、种族或免疫能力以及本领域技术人员将理解的其他因素。给药剂量、数量、频率或持续时间可成比例增加或减少,表现为任何不良副作用、并发症或者治疗或疗法的其他风险因素以及受试者的状态。本领域技术人员将理解可影响提供足以提供治疗或预防利益的量所需的剂量和时间的因素。
可向药物组合物中加入主题aav载体和其它组合物,例如,药学可接受的载体或赋形剂。这样的药物组合物可用于体内或离体施用和递送给受试者等。
本发明所用的术语“药学可接受的”和“生理学可接受的”表示适用于一种或多种施用、体内递送或接触的途径的生理学可接受的制剂,其为气态、液态或固态或其组合。“药学可接受的”或“生理学可接受的”组合物是不会在生理学或其它方面不理想的材料,例如,所述材料可被施用给受试者而不会导致显著的不理想生物学效应。因此,这样的药物组合物可用于例如将病毒载体或病毒颗粒施用给受试者。
这样的组合物包含可与药物施用或体内接触或递送相容的溶剂(水性或非水性)、溶液(水性或非水性)、乳液(例如,水包油型或油包水型)、悬浮液、糖浆、酏剂、分散和悬浮介质、涂层、等渗和吸收促进剂或延迟剂。水性和非-水性的溶剂、溶液和悬浮液可包含悬浮剂和增稠剂。这样的药学可接受的载体包括片剂(带包衣或无包衣的)、胶囊(硬或软的)、微珠、粉末、颗粒和晶体。补充的活性化合物(例如,防腐剂、抗菌剂、抗病毒剂和抗真菌剂)也可加入组合物中。
药物组合物可配制成与具体的施用或递送途径相容,如本发明所述或本领域技术人员所知的。因此,药物组合物包括适用于通过多种途径施用的载体、稀释剂或赋形剂。
适用于肠胃外施用的组合物包含活性化合物的水性和非水性的溶液、悬浮液或乳液,该制剂通常为无菌的,并且可与预期接受者的血液等渗。非限制性的示例性实例包括水、盐水、右旋糖、果糖、乙醇、动物油、植物油或合成油。
还可向制剂中添加助溶剂和助剂。助溶剂的非限制性实例包括羟基或其它极性基团,例如,醇,诸如异丙醇;二醇,诸如丙二醇、聚乙二醇、聚丙二醇、二醇醚;甘油;聚氧乙基脂肪醇醚(polyoxyethylenealcohol)和聚氧乙基脂肪酸酯。助剂包括,例如,表面活性剂,诸如大豆卵磷脂和油酸;脱水山梨糖醇酯,诸如脱水山梨糖醇三油酸酯;和聚乙烯吡咯烷酮。
适合用于本发明的组合物、方法和用途中的药物组合物和递送系统在本领域中是已知的(参见,例如,remington:thescienceandpracticeofpharmacy(2003),第20版,mackpublishingco.,easton,pa;remington’spharmaceuticalsciences(1990),第18版,mackpublishingco.,easton,pa;themerckindex(1996),第12版,merckpublishinggroup,whitehouse,nj;pharmaceuticalprinciplesofsoliddosageforms(1993),technonicpublishingco.,inc.,lancaster,pa.;anselandstoklosa,pharmaceuticalcalculations(2001),第11版,lippincottwilliams&wilkins,baltimore,md;和poznansky等,drugdeliverysystems(1980),r.l.juliano,ed.,oxford,n.y.,pp.253-315)。
本发明所用的“单位剂型”是指适合作为待治疗受试者的单一剂量的物理上独立的单位;每个单位包含经计算在以一个或多个剂量施用时产生所需效果(例如预防或治疗效果)的预定量,其任选地与药物载体(赋形剂、稀释剂、介质或填充剂)结合。单位剂型可置于例如安瓶和小瓶内,所述安瓶和小瓶可包括液体组合物或冷冻干燥或冻干状态的组合物;无菌液体载体,例如,可在施用或体内递送之前添加。各个单位剂型可被包含在多剂量药盒或容器中。aav载体及其药物组合物可被包装在单个或多个单位剂型中以便于施用和剂量均匀性。
在相关实施方案中,本发明所述的aav递送系统可容易地适于递送其它核酸酶(诸如talen或zfn)或rnai构建物(一旦位于靶细胞内其编码和产生功能性sirna)或aso构建物(其编码和产生功能性反义寡核苷酸)至靶细胞(例如,体外、体内或离体的opc或nsc)。
用途
本发明所公开的方法和组合物用于提供髓鞘相关疾病的基于细胞的疗法。细胞可在体内修饰或可离体修饰并随后施用给受试者。在一些实施方案中,将原位编辑患者中枢神经系统中的nsc、opc、神经元细胞和胶质细胞(诸如少突胶质细胞、星形胶质细胞、室管膜细胞和小神经胶质细胞)。在一些实施方案中,细胞将离体修饰并移植到患者体内。
使用受试者自身的细胞消除了对用于移植的供体和受体之间hla匹配的要求。此外,已显示本发明所述的基因修饰细胞适合用于连续(二次)移植,其中可从受试者分离干细胞,并且这些细胞保留了基因修饰并且可被施用给一个或多个其它受试者。因此,所述方法和组合物提供了髓鞘相关障碍的治疗和/或预防。
可使用plp1、plp1的一部分或plp调控元件的靶向缺失来纠正异常基因,在内源性基因中产生功能丢失突变,或者改变内源性基因的表达。在其它方面,可使用plp1或plp1基因调控元件核苷酸序列的抗plp1供体的靶向整合纠正异常基因,插入野生型基因,在内源性基因内产生功能获得突变,或者改变内源性基因的表达。例如,可将编码plp1的转基因或plp1基因调控元件转基因整合到细胞中以提供产生能够增强功能性髓鞘产生的非缺陷蛋白质的细胞(例如,少突胶质细胞或祖细胞)。plp1或plp1基因调控元件的靶向敲除或基因沉默或通过本发明所述方法进行修饰可提供通过经由plp1基因的失活降低plp1毒性来增强功能性髓鞘产生的细胞。基因组编辑还可包括在内源性基因中纠正或引入突变(例如,点突变)来例如修饰内源性plp1基因表达。
可将本发明所述的组合物(例如,细胞和/或核苷酸)施用给受试者以治疗髓鞘相关疾病和障碍。设想通过本发明一些方面治疗的髓鞘相关疾病和障碍可包括与受试者的脱髓鞘、髓鞘形成和髓鞘再生的不足或者髓鞘形成障碍相关的任何疾病、病症(例如,因外伤性脊髓损伤和脑梗塞而发生的那些)或障碍。本发明所用的髓鞘相关障碍可起因于源自多种神经毒性损伤的髓鞘形成相关障碍或脱髓鞘。本发明所用的“脱髓鞘”是指脱髓鞘化或隔绝神经的髓鞘丢失的作用,其是一些神经变性自体免疫疾病的标志,包括多发性硬化、横贯性脊髓炎、慢性炎性脱髓鞘性多发性神经病和格林-巴利综合征。脑白质营养不良是由遗传性酶缺乏引起的,其引起cns白质内髓鞘的异常形成、破坏和/或异常更新。获得性和遗传性髓鞘障碍的预后都很差,导致严重残疾。因此,本发明的一些实施方案可包括治疗受试者的神经变性自体免疫疾病的方法。神经元的髓鞘再生需要少突胶质细胞。本发明所用的术语“髓鞘再生”是指通过更换髓鞘产生细胞或恢复其功能使神经髓鞘再生。
可通过本发明方法治疗或改善的髓鞘相关疾病或障碍包括涉及受试者脑细胞(例如,cns神经元)中的髓鞘形成障碍或脱髓鞘的疾病、障碍或损伤。这样的疾病包括但不限于其中围绕神经元的髓鞘不存在、不完整、没有正确形成或正在恶化的疾病和障碍。这样的疾病包括但不限于多发性硬化(ms)、视神经脊髓炎(nmo)、进行性多灶性脑白质病(pml)、脑脊髓炎(epl)、脑桥中央髓鞘溶解症(cpm)、肾上腺脑白质营养不良、亚历山大病、佩梅病(pmd)、沃勒变性、视神经炎、横贯性脊髓炎、肌萎缩性脊髓侧索硬化症(als)、亨廷顿病、阿尔茨海默病、帕金森病、脊髓受伤、外伤性脑损伤、辐射后损伤、化疗的神经并发症、中风、急性缺血性视神经病、维生素e缺乏症、孤立性维生素e缺乏症综合征、ar、巴-科综合征、马毕二氏综合征、异染性脑白质病变、三叉神经痛、急性播散性脑炎、格林-巴利综合征、marie-charcot-tooth病和贝尔面瘫。
可通过本发明方法治疗或改善的髓鞘相关疾病或障碍包括特征在于髓鞘缺乏的疾病或障碍。在大量神经病学障碍中均涉及了中枢神经系统中的髓鞘形成不足。其中有脑瘫的形式,其中患有脑室周围白质软化的儿童的前脑髓鞘形成的先天性缺陷导致了神经病学发病(goldman等,2008)goldman,s.a.,schanz,s.和windrem,m.s.(2008)。基于干细胞的策略用于治疗小儿髓鞘形成障碍。hummolgenet.17,r76-83。在年龄谱的另一端,髓鞘丢失和无效修复可导致与衰老相关的认知功能下降(kohama等,2011)kohama,s.g.,rosene,d.l.和sherman,l.s.(2011)age(dordr)。人和非人灵长类动物白质的年龄相关变化:从髓鞘紊乱到认知下降。因此,设想增强髓鞘形成和/或髓鞘再生的有效的化合物和方法可以在停止pmd和多种髓鞘相关障碍的疾病进展和恢复功能方面有显著的治疗利益。
在一些实施方案中,可将本发明的组合物施用给并未患和/或不容易患髓鞘相关疾病的受试者以增强或促进髓鞘依赖性过程。在一些实施方案中,可将本发明所述的化合物施用给受试者以促进cns神经元的髓鞘化从而增强认知健康受试者的认知,已知后者是髓鞘相关过程。在一些实施方案中,可将本发明所述的化合物与认知增强(益智)剂联合施用。示例性的试剂包括改善健康个体的认知功能,特别是执行功能、记忆、创造性或积极性的任何药物、补充剂或其它物质。非限制性实例包括西坦类药物(racetams)(例如,吡拉西坦、奥拉西坦和阿尼西坦)、营养保健品(例如,假马齿苋、人参、银杏和gaba)、兴奋剂(例如,安非他命药物、哌甲酯、eugeroics、黄嘌呤和尼古丁)、l-茶氨酸、托卡朋、左旋多巴、阿托西汀和地昔帕明。
本发明的一个特定方面设想了治疗受试者的pmd。方法包括对受试者施用治疗有效量的上文所述的基因修饰细胞或核苷酸组合物。
总剂量将是取决于若干因素的治疗有效量,所述因素包括:受试者的整体健康、受试者的疾病状态、病症的严重性、改善的观察结果以及选定试剂的制剂和施用途径。治疗有效量的确定属于本领域技术人员的能力范围内。确切的制剂、施用途径和剂量可由个体医生根据受试者的病症来选择。
在一些实施方案中,本发明所述的基因修饰细胞或核苷酸组合物可以有效增强受试者的cns神经元的髓鞘产生的量施用:所述增强表现为髓鞘蛋白(例如,mbp)的量与未治疗的cns神经元或受试者的髓鞘蛋白水平相比增加了至少5%、10%、20%、25%、30%、40%、50%、60%、70%、75%、80%、85%、90%、95%、100%、110%、120%、130%、140%、150%、160%、170%、180%、190%、200%、250%、300%、350%、400%、450%、500%、550%、600%、650%、700%、750%、800%、850%、900%、950%或1000%。
在其他实施方案中,基因修饰细胞或核苷酸组合物可以有效促进受试者的cns神经元的存活的量施用,所述促进表现为存活神经元的数量与未经处理的cns神经元或受试者的存活神经元数量相比提高了至少5%、10%、20%、25%、30%、40%、50%、60%、70%、75%、80%、85%、90%、95%、100%、110%、120%、130%、140%、150%、160%、170%、180%、190%、200%、250%、300%、350%、400%、450%、500%、550%、600%、650%、700%、750%、800%、850%、900%、950%或1000%。
治疗罹患髓鞘相关障碍的受试者的另一种策略是施用治疗有效量的本发明所述的基因修饰细胞或核苷酸组合物以及治疗有效量的少突胶质细胞分化和/或增殖诱导剂和/或抗神经变性病试剂。抗神经变性疾病试剂的实例包括左旋多巴、胆碱酯酶抑制剂、抗胆碱能药、多巴胺激动剂、类固醇和免疫调节剂(包括干扰素、单克隆抗体和醋酸格拉替雷)。
因此,在本发明的进一步的方面,本发明所述的基因修饰细胞或核苷酸组合物可作为与神经变性和髓鞘相关障碍的辅助疗法的组合治疗的一部分来施用。
短语“组合治疗”包括施用本发明所述的少突胶质细胞前体分化诱导化合物和治疗剂作为特定治疗方案的一部分,所述治疗方案旨在通过这些治疗剂的共同作用提供有益效果。当组合施用时,少突胶质细胞前体分化诱导化合物和治疗剂可被配制为单独的组合物。这些治疗剂的组合施用通常在限定时间段进行(通常为分钟、小时、日或周,取决于所选择的组合)。
“组合治疗”旨在包含以顺序方式施用这些治疗剂,即,其中每种治疗剂在不同时间施用,并且这些治疗剂或至少两种治疗剂以基本同时的方式施用。基本同时施用可通过例如对受试者施用具有固定比率的每种治疗剂的单个胶囊或者每种治疗剂的多个单一胶囊而实现。每种治疗剂的顺序或基本同时施用可通过任何合适途径进行,包括但不限于口服途径、静脉内途径、肌肉内途径和通过粘膜组织直接吸收。治疗剂可通过相同途径或通过不同途径施用。例如,组合的第一种治疗剂可通过静脉注射施用,而组合的另一种治疗剂可口服施用。或者,例如,所有治疗剂可口服施用或者所有治疗剂可通过静脉注射施用。治疗剂的施用顺序并不是非常严格的。“组合治疗”也可包含如上所述的治疗剂进一步与其他生物活性成分(诸如但不限于,第二种不同治疗剂)和非药物疗法(例如,手术)组合施用。
在本发明的另一方面,与本发明所述的基因修饰细胞或核苷酸组合物组合治疗施用的治疗剂可包括至少一种抗神经变性剂,诸如但不限于免疫治疗剂。
用于本发明方法中的免疫治疗剂可包括靶向疾病的免疫组成部分和/或在缓解-复发性髓鞘相关障碍(如多发性硬化)的急性发作期中证实的急性炎性反应的疗法。实例包括但不限于免疫调节剂,诸如干扰素β-la和β-lb(分别为avonex和betaseron)、那他珠单抗(copaxone)那他珠单抗(tysabri)、醋酸格拉替雷(copaxone)或米托蒽醌。
通过以下实施例进一步举例说明本发明,其并非旨在限制权利要求的范围。
实施例1:核酸酶介导的plp1失活以治疗遗传学髓鞘障碍
介绍
髓鞘形成异常导致异常的神经元信号传导和神经功能障碍。通过纠正其内源性髓鞘产生细胞中的基因异常来恢复这些患者的髓鞘产生能力代表了有前景的治疗途径,然而这种治疗方法的可行性尚需验证。因此,我们选择了通过关注一种被称为佩梅病(pmd)的严重的原型脑白质营养不良来验证这一范例。
pmd是由蛋白脂蛋白1(plp1)中的突变导致的严重的x-连锁遗传的髓鞘障碍。pmd患者通常经历严重的神经疾病,包括严重的认知和运动障碍,其不可避免地在儿童期或青春期的早期死亡中达到高峰。因此,我们期望确定是否可通过在plp1基因或其调控元件中引入核酸酶介导的插入或缺失(插入缺失)来治疗严重的pmd患者,所述插入缺失能有效和永久性地失活或降低plp1表达(plp1-插入缺失)。核酸酶介导的插入缺失可靶向少突胶质细胞或其祖细胞,包括神经干细胞或少突胶质细胞前体细胞(opc)。靶向祖细胞将提供永久和自我放大的治疗,因为plp-插入缺失祖细胞将在患者的整个生命周期上持续产生功能性plp1-插入缺失的少突胶质细胞。鉴于已经在患者中鉴定了许多不同和各种pmd-致病突变,我们的plp1-插入缺失方法是独特的,因为其提供了适用于所有pmd患者的通用方法。
jimpy小鼠
为开发plp1-插入缺失治疗,我们利用了名为jimpy的pmd小鼠模型。这种小鼠在plp1基因的内含子4的剪接受体位点中具有点突变,导致错误折叠的plp1蛋白,其最终导致少突胶质细胞丢失、严重的髓鞘形成减少、严重的震颤、共济失调、癫痫发作和出生后第三周之前的早期死亡。因此,其有效地概括了人类患者中可见的严重pmd的许多方面。
jimpy小鼠代表最严重的pmd模型。图2是显示小鼠plp1基因中jimpy基因突变的位置以及最终导致少突胶质细胞死亡的所得基因产物的示意图(不按比例)。该图下部显示,jimpy小鼠的寿命严重缩短,平均存活仅为约23日。
图3显示了jimpy小鼠脑中的严重的髓鞘形成减少。注意,与野生型对照物相比,p19(出生后19日)jimpy小鼠脑部中的mbp染色显著减少。在相同年龄,jimpy小鼠还显示了意向震颤和共济失调(数据未示出)。
向导rna选择和验证
crispr-cas9是通过引入核酸酶cas和22bp单一向导rna(sgrna)而在哺乳动物细胞中诱导插入缺失的有效的核酸酶-介导系统的一个实例。合适的sgrna如以下文献中所述设计:hsu等(2013)naturebiotechnology31,827-832。具体地,选择sgrna以最大化在靶(on-target)和最小化脱靶(off-target)的插入缺失形成以及早期外显子靶向,以提高无义介导衰变的效率。通过clontech’sguide-itsgrna筛选试剂盒验证sgrna的核酸酶活性,和通过使用thermofisherneon转染系统将表达sacas9或spcas9与sgrna的质粒电穿孔至小鼠和人细胞中来验证功能性插入缺失的诱导。切割效率和sgrna分级通过如下方式确定:使用illummamiseq系统深度测序来量化插入缺失形成。最佳sgrna在表1中详述。
表1:最佳验证的sgrna序列
图4和5是显示通过靶向外显子3的crisprspcas9/双向导rna(sgrna)敲除jimpy小鼠的受精卵中的plp1基因以产生cr-impyko后代小鼠的示例性方法的示意图。图4显示了外显子3中sgrnaa和b靶向位点的相对位置。图5显示了产生用于接收sgrna和spcas9mrna的jimpy雄性受精卵的一般实验方法。成功的crispr/cas9介导的jimpyplp1基因的敲除导致由代孕宿主雌性娩出的plp1为空的雄性cr-impy初代的诞生。与亲本体杂交两代得到了用于进一步表征的子代小鼠。
动物中spcas9-介导的plp1缺失
作为我们的治疗性plp1-插入缺失策略的初始概念验证,将来自jimpy育种者的受精卵用200ng/μlspcas9mrna和10ng/μl的每种sgrnaa和b电镀,然后将其植入代孕雌性中(参见图5)。这导致在plp1中外显子3的5'末端上携带~80个核苷酸缺失的crispr敲除的jimpy(cr-impy[或crimpy])雄性小鼠的产生。虽然jimpy雄性表现出严重的表型并且在出生后的第三周死亡,但是该crimpy雄性与jimpy雄性完全不同,因为它没有表现出震颤、癫痫发作、共济失调或早期死亡(数据未显示)。值得注意的是,这种crimpy雄性能够成功繁殖和生存超过六个月,然后将其处死进行组织学分析,证明中枢神经系统的完全髓鞘化与野生型无法区分(见图1)。将crimpy等位基因传递给健康的孙代证实了我们的plp1-插入缺失治疗的显著治疗效果。
事实上,图6显示了cr-impy小鼠与jimpy和野生型对照物相比寿命有所恢复(每组n>15,p<0.0001)。
图7显示,通过检测mbp的全脑ihc,cr-impy小鼠显示了在成熟少突胶质细胞中的恢复。在出生后第19日,野生型和cr-impy小鼠具有比jimpy小鼠显著更多的mbp染色。在出生后6个月,虽然所有的jimpy小鼠都已死亡,但野生型和cr-impy小鼠的mbp染色水平难以区分。
还使用了两种功能性测试来评估cr-impy小鼠中的运动协调和自主活动。
图8示出了用于评估cr-impy小鼠的运动协调的旋转杆测试的示意图,其中通过在旋转杆加速时从旋转杆下落的测量时间来定量运动协调。在出生后19日、2个月和6个月,在每个时间点,对野生型、jimpy和cr-impy小鼠进行测量。不同值之间的统计显著性由p值表示。在p19,与jimpy小鼠相比,野生型和cr-impy小鼠均具有在统计学上显著更长的下落时间。在出生后2个月和6个月,所有的jimpy小鼠均已死亡,而野生型和cr-impy小鼠的测得的下落时间水平难以区分。结果显示,与野生型和jimpy小鼠相比,cr-impy小鼠的运动协调有所恢复。
图9显示了用于评估cr-impy小鼠的自主活动的旷场测试的示意图,其中用通过自动视频跟踪进行5分钟跟踪时测得的在盒子中行进的总距离来定量自主活动。在出生后的p19、2个月和6个月,在每个时间点,在野生型、jimpy小鼠和cr-impy小鼠中进行测量。不同值之间的统计显著性由p值表示。在p19,与jimpy小鼠相比,野生型和cr-impy小鼠的总行进距离在统计学上显著更长。在出生后2个月和6个月,虽然所有的jimpy小鼠都已经死亡,但野生型和cr-impy小鼠中测量的总行进距离水平难以区分。结果显示,与野生型和jimpy小鼠相比,cr-impy小鼠的自主活动恢复。
基于轴突刺激后传导速度的测量进行进一步的功能测试。图10显示了视神经传导速度测试的示意图以及较快和较慢传导峰的代表性结果–分别为第一峰和第二峰。与无髓鞘和较小直径的轴突相比,有髓鞘和大直径轴突通常具有更快的传导。在出生后d19,分别通过第1和第2峰测量快和慢传导速度,其在野生型、jimpy小鼠和cr-impy小鼠中任两个之间均在统计学上显著不同。然而,在出生后6个月,虽然所有的jimpy小鼠都已死亡,但是野生型和cr-impy小鼠之间的差异(如果有的话)在统计学上是不显著的。这表明cr-impy小鼠的轴突传导速度速度虽然最初落后于野生型小鼠,但最终会随着时间的推移而逐渐增加。
实际上,在约6月龄时,野生型和cr-impy小鼠视神经的电子显微镜(em)成像显示没有可辨别的差异。参见图11。
cr-impy小鼠在6月龄时继续显示与野生型类似的功能性性能,显示了基因治疗介导的纠正的长期功能性稳定性。
这些结果证明向plp1中引入失活的插入缺失可完全拯救pmd小鼠模型。
总结
本发明涉及产生基因修饰细胞的方法及其用于治疗人髓鞘障碍的使用方法。具体地,我们使用了蛋白脂蛋白1(plp1)基因的核酸酶-介导的基因破坏作为髓鞘相关障碍的新型治疗方法,并在名为佩梅病(pmd)的脑白质营养不良的背景下验证了功效。
图12是显示本发明一个实施方案的示意图,其中将crispr-cas9介导的基因沉默递送至出生后的脑中,例如通过编码sacas9和sgrna的aav病毒载体,其可以至少部分地纠正患者脑中的突变opc。如此纠正的opc将及时胜过任何突变opc,从而提供充分恢复的髓鞘形成和患者神经元功能。
我们的策略通过plp1基因的失活绕过plp1相关的毒性,这增强了生产功能性髓鞘的能力。在一些情况下,可以原位编辑患者中枢神经系统中的神经干细胞、少突胶质细胞祖细胞(opc)或其他神经胶质细胞。在另一个例子中,细胞可以离体修饰并移植到患者体内。
更具体地,我们已证明了使用在plp1基因或plp1基因调控元件中核酸酶介导的插入缺失的产生、以分别使plp1翻译或转录失活的新治疗策略。我们已示出,啮齿动物体内核酸酶介导的plp1基因编辑恢复了患有严重pmd的小鼠的正常功能和完整寿命。我们证明了plp1的这种扰动在体外恢复了pmd模型细胞的功能。作为这些概念验证研究的一部分,示例了具有定点向导rna的crispr-cas9核酸酶,但是该治疗策略可以包括任何定点核酸酶或基因编辑技术。
由于在患者中看到的多种dna突变谱,pmd和其他脑白质营养不良患者的治疗方法的开发具有挑战性。在这里,我们为所有pmd患者提供通用治疗。该单一方法/产品可用于所有pmd患者,以有效失活有害的突变plp1基因。此外,plp1基因的失活可有益于其他髓鞘疾病,以减轻少突胶质细胞中的细胞应激。因此,该治疗方法具有相当大的价值,并且在追求plp1失活临床疗法方面具有强大的药学和经济意义。
应当理解,本发明描述的方法可以以多种方式并且通过本领域公知的各种修改和置换来实施。还可以理解,关于作用模式提出的任何理论不应被解释为以任何方式限制本发明,而是呈现为使得可以更全面地理解本发明的方法。
上述说明书中提及的所有出版物和专利均通过引用结合到本发明中。
实施例2使用crispr/cas9的aav递送的plp1的出生后失活
该实施例证明plp1的出生后失活可用于有效治疗pmd或降低pmd的严重性。
为有利于将具有靶向plp1基因座的sgrna的crispr/cas9系统递送到患者中,生产了数种靶向cns的aav血清型,包括php.b,并且验证了对于opc的aav趋向性。aav构建物(aav9或aav-php.b)含有sacrispr-cas9核酸酶(cmv-sacas9,cmv启动子控制下的sacas9编码序列),其具有针对plp1的定点向导rna(sgrna)(u6-sgrna,sgrna在u6启动子的控制下),其被设计为在plp1基因中产生插入缺失,从而阻止有缺陷的plp1蛋白的表达。
根据casewesternreserveuniversity的机构动物管理及使用委员会审查批准的方案饲养小鼠。将小鼠置于温度和湿度受控的居住单元中,每天12小时/光照周期,并允许随意获取食物。
然后通过立体定位向心室内注射用含有crispr的aav或编码gfp的对照处理jimpy(严重的佩梅病小鼠模型)小鼠。具体而言,从jimpy育种配对获得出生后0日的雄性幼崽,并使用冷冻麻醉快速麻醉。
将带有32号针头的10μlhamilton注射器中装入包装了cmv-sacas9和靶向plp1的u6-sgrna的aav(aav9或aav-php.b)。将针头从矢状缝和人字缝与眼睛的交叉点的2/5处下降通过颅骨至约2mm的深度。将2μl病毒溶液注入侧脑室。然后使用相同的坐标在对侧的侧脑室重复注射,并且向心室系统总共输送约1×1010-1×1011个载体基因组的注射体积。对照小鼠重复相同的操作。
允许幼崽在加热垫上恢复,然后重新送回给其母亲。每天监测幼崽与未处理或用介质处理的jimpy动物相比的表型改善,所述未处理或用介质处理的jimpy动物在2周龄时发展出严重的运动表型(例如,意向震颤和癫痫发作),并且在3周龄时死亡。
使用行为(例如,用于运动表现的旋转棒和旷场测试,参见实施例1和图8和9)、组织学(用于髓鞘蛋白的cns免疫染色和用于髓鞘超结构的电子显微镜,参见实施例1和图7和11)分析存活超过3周的经处理的动物,或每日监测寿命延长的统计分析(参见实施例1和图6)。
此外,确定cns中需要编辑的细胞数量的空间要求(基因编辑细胞的“剂量反应”)以通过免疫组化产生功能性反应。
最后,通过在出生后p1日、p7日和p14日引入优化构建物来探索时间关系。
经优化的限定治疗窗口内的crispr/cas9方法降低了突变plp1蛋白的表达,增加了治疗个体的寿命,并恢复了轴突的髓鞘形成。
实施例3使用反义寡核苷酸(aso)的plp1的出生后敲减
该实施例证明了使用aso在出生后下调或敲减plp1基因活性可用于有效治疗pmd或降低pmd的严重性。
根据casewesternreserveuniversity的机构动物管理及使用委员会审查批准的方案饲养小鼠。将小鼠置于温度和湿度受控的居住单元中,每天12小时/光照周期,并允许随意获取食物。
从jimpy(严重的佩梅病小鼠模型)育种配对获得出生后0日的雄性幼崽,并使用冷冻麻醉快速麻醉。将带有32号针头的10μlhamilton注射器中装入靶向plp1的反义寡核苷酸。将针头从矢状缝和人字缝与眼睛的交叉点的2/5处下降通过颅骨至约2mm的深度。将2μlaso溶液注入侧脑室。然后使用相同的坐标和注射体积在对侧的侧脑室重复注射,以向心室系统输送约10-75μgaso。
允许幼崽在加热垫上恢复,然后重新送回给其母亲。每天监测幼崽与未处理或用介质处理的jimpy动物相比的表型改善,所述未处理或用介质处理的jimpy动物在2周龄时发展出严重的运动表型(例如,意向震颤和癫痫发作),并且在3周龄时死亡。
使用行为(例如,用于运动表现的旋转棒和旷场测试,参见实施例1和图8和9)、组织学(用于髓鞘蛋白的cns免疫染色和用于髓鞘超结构的电子显微镜,参见实施例1和图7和11)分析存活超过3周的经处理的动物,或每日监测寿命延长的统计分析(参见实施例1和图6)。
实施例4使用rnai在出生后敲减plp1
该实施例证明使用rnai在出生后下调或敲低plp1基因活性可用于有效治疗pmd或降低pmd的严重性。
根据casewesternreserveuniversity的机构动物管理及使用委员会审查批准的方案饲养小鼠。将小鼠置于温度和湿度受控的居住单元中,每天12小时/光照周期,并允许随意获取食物。
从jimpy(严重的佩梅病小鼠模型)育种配对获得出生后0日的雄性幼崽,并使用冷冻麻醉快速麻醉。将带有32号针头的10μlhamilton注射器中装入包装了靶向plp1的cmv-rnai(一种rnai构建体,其可在cmv启动子的控制下在细胞内转录以产生功能性rnai分子,该分子然后可以加工成靶向plp1的sirna/shrna/mirna)的aav(aav9或aav-php.b)。或者,使用非病毒sirna载体,例如细胞穿透肽、聚合物、树枝状大分子、sirna生物缀合物和基于脂质的sirna载体等,针头装载靶向plp1的rnai构建体的非病毒制剂。将针头从矢状缝和人字缝与眼睛的交叉点的2/5处下降通过颅骨至约2mm的深度。将2μlrnai溶液注入侧脑室。然后使用相同的坐标和注射体积在对侧的侧脑室中重复注射。
允许幼崽在加热垫上恢复,然后重新送回给其母亲。每天监测幼崽与未处理或用介质处理的jimpy动物相比的表型改善,所述未处理或用介质处理的jimpy动物在2周龄时发展出严重的运动表型(例如,意向震颤和癫痫发作),并且在3周龄时死亡。
使用行为(例如,用于运动表现的旋转棒和旷场测试,参见实施例1和图8和9)、组织学(用于髓鞘蛋白的cns免疫染色和用于髓鞘超结构的电子显微镜,参见实施例1和图7和11)分析存活超过3周的经处理的动物,或每日监测寿命延长的统计分析(参见实施例1和图6)。
-
 0135587... 来自[中国] 2022年02月06日 23:03请问这个技术国内有吗?国外成熟吗能不能看好佩梅病啊?谢谢
0135587... 来自[中国] 2022年02月06日 23:03请问这个技术国内有吗?国外成熟吗能不能看好佩梅病啊?谢谢