一种稳定同位素标记的磺胺甲恶唑及其合成方法与流程
本发明涉及同位素标记领域,特别是涉及一种稳定同位素标记的磺胺甲恶唑及其合成方法。
背景技术:
磺胺甲恶唑,化学名为n-(5-甲基-3-异噁唑基)-4-氨基苯磺酰胺,为白色结晶性粉末;无臭,味微苦。在水中几乎不溶;在稀盐酸、氢氧化钠试液或氨试液中易溶。磺胺甲恶唑属于磺胺类药,主要用于治疗敏感菌引起的尿路感染、呼吸系统感染、肠道感染、胆道感染及局部软组织或创面感染等。
磺胺类药物临床上主要用于预防和治疗感染性疾病,加之其性质稳定,制造不需粮食做原料、产量大、品种多、价格低、使用简便、供应充足等优点,兽医临床和畜牧养殖业中作为饲料添加剂或动物疾病治疗药物广泛应用。但是磺胺药会引起人过敏性反应,且可能有致癌性,随着社会的发展,磺胺类药物的不合理使用,使其在动物性食品中残留引起生态环境污染和人类健康危害的潜在威胁已备受关注,成为人类亟待解决的问题之一,而各类检测方法也随之应运而生。
传统的食品安全磺胺类药物残留检测方法主要有高效液相色谱法(hplc)、液相色谱-质谱法(hplc/ms)、薄层色谱法(tlc)、气相色谱法(gc)、气相色谱-质谱联机(gc/ms)、毛细管电泳(ce)等,但是这些方法普遍存在灵敏度低,方法繁琐,或只能进行半定量分析等缺点。
稳定同位素稀释质谱法idms(isotopedilutionmassspectrometry)是采用与被测物质具有相同分子结构的稳定同位素标记的化合物作为内标物质,用高分辨液相色谱-质谱联用仪(lc-ms)进行检测,通过质谱仪测量相应质量数的离子的比值并与标准的比值比较达到准确定量的目的。采用同位素内标可以有效消除样品在化学和物理的前处理步骤中所引起的回收率差异,从而避免处理样品过程中的损失对检测结果造成的偏差。
稳定同位素内标的特性与lc-ms的高灵敏度和处理复杂样品的能力结合,使得色谱-同位素稀释质谱技术被公认为测量微量及痕量有机物的基准定量方法,应用越来越广泛。而由于同位素标记原料价格较贵,成本较高、生产工艺较难实现、如何保证生产过程同位素原子不会脱落、产品纯化等技术难点,稳定同位素标记的磺胺甲恶唑-d4的合成方法尚未见报道,这严重制约了我国食品安全领域兽药残留检测行业的发展。因此,有必要提供一种稳定同位素标记的磺胺甲恶唑-d4的合成方法,能得到化学纯度和同位素丰度均非常高的磺胺甲恶唑-d4,这将为定量检测食品残留物磺胺甲恶唑提供标准试剂,从而有效的为我国食品安全领域兽药残留检测服务。
技术实现要素:
本发明所要解决的技术问题是提供一种稳定同位素标记的磺胺甲恶唑及其合成方法,可以作为定量检测磺胺甲恶唑的标准试剂;且制备过程简单,产品容易分离提纯,得到的产品化学纯度高。
本发明为解决上述技术问题而采用的技术方案是提供一种稳定同位素标记的磺胺甲恶唑的合成方法,包括以下步骤:s1:将稳定同位素标记的苯胺与乙酸酐进行反应,制得稳定同位素标记的乙酰氨基苯;s2:将稳定同位素标记的乙酰氨基苯与氯磺酸进行反应,制得稳定同位素标记的对乙酰氨基苯磺酰氯;s3:将稳定同位素标记的对乙酰氨基苯磺酰、3-氨基-5甲基异恶唑以及4-二甲氨基吡啶进行反应,制得稳定同位素标记的醋磺胺甲恶唑;s4:将稳定同位素标记的醋磺胺甲恶唑在碱性条件下水解,制得稳定同位素标记的磺胺甲恶唑。
进一步地,所述步骤s1过程如下:按重量份数计,惰性气体保护下,在反应容器中加入1份稳定同位素标记的苯胺,逐滴滴加2~3份乙酸酐,25~30℃反应2~5小时,加入6~8份水,饱和碳酸氢钠溶液中和,二氯甲烷萃取,有机相饱和食盐水洗涤,去除溶剂后得到稳定同位素标记的乙酰氨基苯。
进一步地,所述滴加2~3份乙酸酐是在冰盐浴下进行,温度保持在0~5℃。
进一步地,所述步骤s2过程如下:按重量份数计,惰性气体保护下,在反应容器中加入1份稳定同位素标记的乙酰氨基苯,逐滴滴加8~10份氯磺酸,25~30℃反应2~5小时,加入6~8份水,析出固体,过滤,滤饼水洗,滤饼在70~80℃干燥2~3小时,得到稳定同位素标记的对乙酰氨基苯磺酰氯。
进一步地,所述滴加8~10份氯磺酸是在冰盐浴下进行,温度保持在0~5℃;所述加入6~8份水中的水为冰水,且在冰盐浴下进行,缓慢加入,温度保持在0~10℃。
进一步地,所述惰性气体为氮气或氩气。
进一步地,所述步骤s3过程如下:按重量份数计,在反应容器中依次加入1份稳定同位素标记的对乙酰氨基苯磺酰氯,0.4~0.5份3-氨基-5甲基异恶唑,0.02~0.03份4-二甲氨基吡啶,再加入10~15份溶剂,25~30℃反应10~15小时,减压蒸馏去除溶剂,水稀释,用乙酸乙酯萃取,有机相水洗、酸洗,浓缩后重结晶得到稳定同位素标记的醋磺胺甲恶唑。
进一步地,所述溶剂为干燥无水的吡啶;所述酸洗采用的酸为1m盐酸。
进一步地,所述步骤s4过程如下:按重量份数计,在反应容器中依次加入1份稳定同位素标记的醋磺胺甲恶唑,20~25份溶剂,1.5~2.5份碱,回流反应4~6小时,加入0.1~0.2份脱色剂脱色1~2小时,过滤,滤液在70~80℃滴加酸调节ph至5.0~6.0,过滤,滤饼水洗,滤饼在70~80℃干燥2~3小时,得到稳定同位素标记的磺胺甲恶唑。
进一步地,所述溶剂为水;所述碱为氢氧化钠;所述脱色剂为活性炭;所述酸为2m盐酸。
本发明为解决上述技术问题而采用的另一技术方案是提供一种稳定同位素标记的磺胺甲恶唑,由上述合成方法获取,具有如下所示的分子结构:
本发明具有如下有益效果:本发明提供的稳定同位素标记的磺胺甲恶唑及其合成方法,以同位素丰度在99%atom以上的苯胺-d5为同位素标记来源,通过简单的四步反应合成磺胺甲恶唑-d4,每一步反应过程氘原子均不会脱落,稳定同位素原子利用率高;且本发明合成过程简单,产品容易分离提纯,得到的产品化学纯度与同位素丰度均达到99%以上,满足作为定量检测磺胺甲恶唑的标准试剂的要求;使用价值高、具有良好的经济性。
附图说明
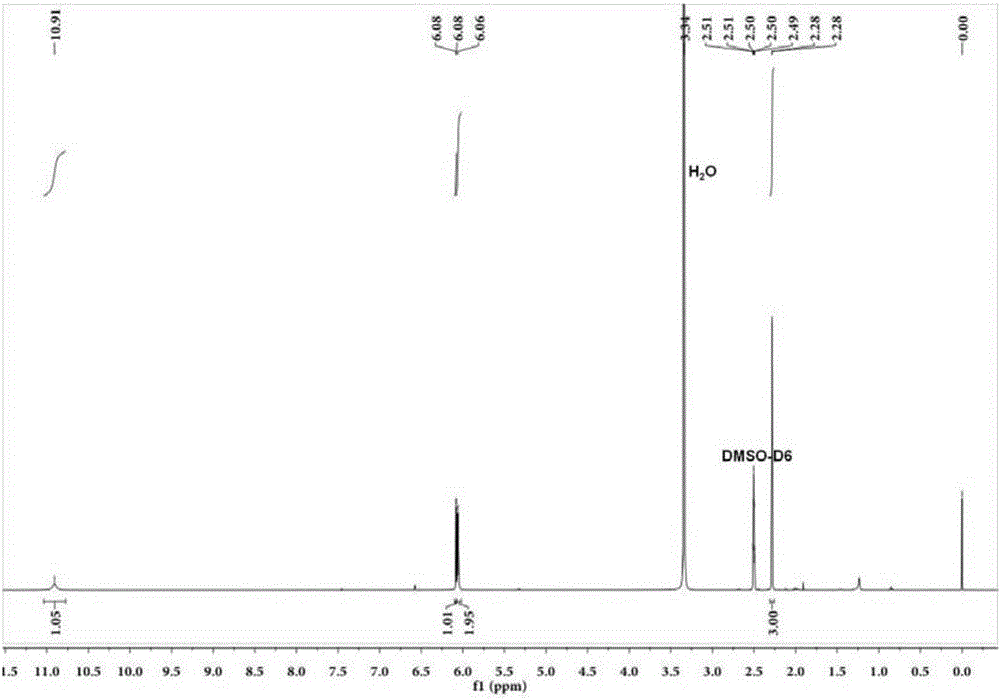
图1为实施例1得到的稳定同位素标记的磺胺甲恶唑-d4的核磁共振氢谱图。
图2为实施例1得到的稳定同位素标记的磺胺甲恶唑-d4的高效液相色谱图。
具体实施方式
下面结合附图和实施例对本发明作进一步的描述,但不应理解为是对本发明的限制。
本发明提供的稳定同位素标记的磺胺甲恶唑的合成方法,包括以下步骤:
s1:将稳定同位素标记的苯胺与乙酸酐进行反应,制得稳定同位素标记的乙酰氨基苯;所述稳定同位素标记的苯胺的分子结构为:
所述的稳定同位素标记的乙酰氨基苯的分子结构为:
s2:将稳定同位素标记的乙酰氨基苯与氯磺酸进行反应,制得稳定同位素标记的对乙酰氨基苯磺酰氯;所述对乙酰氨基苯磺酰氯的分子结构为:
s3:将稳定同位素标记的对乙酰氨基苯磺酰、3-氨基-5甲基异恶唑以及4-二甲氨基吡啶进行反应,制得稳定同位素标记的醋磺胺甲恶唑;所述稳定同位素标记的醋磺胺甲恶唑的分子结构为:
s4:将稳定同位素标记的醋磺胺甲恶唑在碱性条件下水解,制得稳定同位素标记的磺胺甲恶唑。所述稳定同位素标记的磺胺甲恶唑的分子结构为:
实施例1
稳定同位素标记的磺胺甲恶唑的分子结构如下:
通过如下合成步骤制取:
s1.冰盐浴、氮气保护下,在反应容器中加入1.76g苯胺-d5,逐滴滴加4.5g乙酸酐,25~30℃反应2.5小时,加入12g水,饱和碳酸氢钠溶液中和,二氯甲烷萃取,有机相饱和食盐水洗涤,有机相去除溶剂后得到乙酰氨基苯-d5,产率99%;
s2.冰盐浴、氮气保护下,在反应容器中加入2.4g乙酰氨基苯-d5,逐滴滴加21.4g氯磺酸,25~30℃反应3小时,重新置于冰盐浴下,加入15g冰水,析出固体,过滤,滤饼水洗,滤饼在70~80℃干燥2~3小时,得到对乙酰氨基苯磺酰氯-d4,产率69%;
s3.在反应容器中依次加入2.0g对乙酰氨基苯磺酰氯-d4,880mg3-氨基-5甲基异恶唑,52mg4-二甲氨基吡啶,再加入20g新干燥的吡啶,25~30℃反应12小时,减压蒸馏去除溶剂,水稀释,乙酸乙酯萃取,有机相水洗、1m盐酸洗涤,有机相浓缩后重结晶,得到醋磺胺甲恶唑-d4,产率60%;
s4.在反应容器中依次加入1.5g醋磺胺甲恶唑-d4,30g水,2.5g氢氧化钠,回流反应4小时,加入150mg活性炭脱色1小时,过滤,滤液在70~80℃滴加2m盐酸调节ph至5.0~6.0,过滤,滤饼水洗,滤饼在70~80℃干燥2~3小时,得到磺胺甲恶唑-d4,产率93%,所得产品的化学纯度以及同位素丰度均达到99%以上。
本实施例获取的产品以dmso-d6为溶剂,通过bruke-400m核磁共振仪器检测得到图1所示的核磁共振氢谱图,从图1可以看出,化学位移~6.5ppm和~7.4ppm处未见吸收峰,表明结构为磺胺甲恶唑-d4。
同时,本实施例获取的产品样品溶于0.1m盐酸,以乙腈:0.5%磷酸=35:65为流动相,1.0ml/min的流速通过柱温为30℃的液相柱(cnw:athenac18-wp4.6*250mm,5um(laeq-462572)),通过dad(205nm)检测器获得磺胺甲恶唑-d4的高效液相色谱图,如图2所示,从图2可以看出,样品纯度达到99%以上。
实施例2
稳定同位素标记的磺胺甲恶唑的合成过程如下:
s1.冰盐浴、氩气保护下,在反应容器中加入2.0g苯胺-d5,逐滴滴加5.0g乙酸酐,25~30℃反应3小时,加入15g水,饱和碳酸氢钠溶液中和,二氯甲烷萃取,有机相饱和食盐水洗涤,有机相去除溶剂后,得到乙酰氨基苯-d5,产率98%;
s2.冰盐浴、氩气保护下,在反应容器中加入2.8g乙酰氨基苯-d5,逐滴滴加26g氯磺酸,25~30℃反应4小时,重新置于冰盐浴下,加入20g冰水,析出固体,过滤,滤饼水洗,滤饼在70~80℃干燥2~3小时,得到对乙酰氨基苯磺酰氯-d4,产率72%
s3.在反应容器中依次加入3.0g对乙酰氨基苯磺酰氯-d4,1.3g3-氨基-5甲基异恶唑,80mg4-二甲氨基吡啶,再加入40g新干燥的吡啶,25~30℃反应14小时,减压蒸馏去除溶剂,水稀释,乙酸乙酯萃取,有机相水洗、1m盐酸洗涤,浓缩后重结晶,得到醋磺胺甲恶唑-d4,产率65%;
s4.在反应容器中依次加入2.0g醋磺胺甲恶唑-d4,50g水,5g氢氧化钠,回流反应6小时,加入300mg活性炭脱色2小时,过滤,滤液在70~80℃滴加2m盐酸调节ph至5.0~6.0,过滤,滤饼水洗,滤饼在70~80℃干燥2~3小时,得到磺胺甲恶唑-d4,产率95%,所得产品的化学纯度以及同位素丰度均达到99%以上。
虽然本发明已以较佳实施例揭示如上,然其并非用以限定本发明,任何本领域技术人员,在不脱离本发明的精神和范围内,当可作些许的修改和完善,因此本发明的保护范围当以权利要求书所界定的为准。
- 还没有人留言评论。精彩留言会获得点赞!