一种利用CRISPRCas9技术纠正猪KIT基因结构突变的方法与流程
本发明属于生物技术领域,具体来说,涉及一种利用crisprcas9技术纠正猪kit基因结构突变的方法。
背景技术
kit基因是一种和猪毛色相关的显性白基因,在染色体上以450kb为单位呈拷贝数变异,并且突变型拷贝在内含子17第一个碱基上存在g>a剪接突变,产生17外显子缺失型kit蛋白。目前仅发现在大白猪和长白猪kit基因中出现这种突变,被认为是控制大白猪和长白猪显性白的原因。
kit基因控制毛色的作用机理是通过c-kit与其配体scf相互作用影响黑色素细胞前体物的迁移和存活,从而决定猪的毛色。kit基因存在三种等位基因,显性等级为i>ip>i,其中i对应完全显性白毛色(大白猪、长白猪),这种基因型含有多个kit基因拷贝,除了带有正常kit基因(kit1)外,还携带两种突变的全长拷贝(kit2);ip表现为白色或有色毛的斑块或斑点性状(皮特兰),其含有两个正常kit基因,导致kit基因的表达量上升,进而影响了scf的可利用性,扰乱了黑色素细胞前体物迁移与存活,致使斑块或斑点表型产生;i等位基因为野生型kit基因,其表型为野灰色毛色(欧洲野猪)。kit基因的结构突变除了影响色素生成外,还影响动物造血系统的发育。小鼠kit基因突变纯合子通常会因红细胞、巨核细胞和肥大细胞的发育缺陷而发生严重的贫血症致死或亚致死,而i/i纯合子仔猪在出生后的第一周内,红细胞数量会显著减少,同时红细胞比容和平均红细胞体积也有显著性的下降。
cas9和sgrna是crispr/cas9系统的基本成分,sgrna用于特异位点识别,cas9用于切割靶位点dna。dna片段的删除通常依赖于一对sgrna作用于目的片段两侧产生两个dsb(双链dna断裂),通过nhej(非同源末端连接)修复来删除插入的dna片段。
技术实现要素:
本发明的目的是提供一种利用crispr/cas9技术纠正猪kit基因结构突变的方法,该方法简单易行,通过能够有效靶向,通过nhej修复方式直接连接,实现拷贝数的精确删除。
为了实现上述目的,本发明采用了以下技术方案。
一种利用crispr/cas9技术纠正猪kit基因结构突变的方法,包括通过crispr/cas9技术构建分别靶向猪kit基因内含子16和内含子17的打靶载体,转染猪肾细胞,使猪kit基因实现拷贝删除,其中构建载体所用的sgrna的核苷酸序列如seqidno.1、seqidno.2、seqidno.3、seqidno.4中的任一项所示。
根据本发明的方法,其具体步骤包括:
1)针对猪kit基因内含子16和内含子17设计sgrna,将变性退火及磷酸化的sgrna双链寡核苷酸片段通过酶切和连接插入到荧光素酶报告基因载体中,转染猪肾细胞,初步筛选sgrna活性;
2)细胞经培养后使用流式细胞仪进行阳性筛选,分离包含荧光报告基因信号的细胞群;
3)qpcr鉴定单细胞克隆kit基因型拷贝数变化,t-a克隆测序检测剪接突变位点是否已被删除。
根据本发明的方法,所述步骤2)中,细胞培养48小时后,利用流式细胞仪进行细胞分选。
根据本发明的方法,所述猪优选但不限于大白猪。
本发明还提供了一种利用crispr/cas9技术纠正猪kit基因结构突变的基因编辑猪的制备方法。
本发明提供的制备方法,为将上述方法制备的含有已纠正猪kit基因结构突变的单细胞克隆通过体细胞核移植获得kit基因编辑猪。
具体地,该方法包括通过crispr/cas9技术构建分别靶向猪kit基因内含子16和内含子17的打靶载体,转染猪肾细胞,使猪kit基因实现拷贝删除,再将含有已纠正猪kit基因结构突变的单细胞克隆通过体细胞核移植获得kit基因编辑猪,其中构建所述打靶载体所用的sgrna的核苷酸序列如seqidno.1、seqidno.2、seqidno.3、seqidno.4中的任一项所示。
根据本发明的方法,所述猪为大白猪。
本发明还提供了一种用于特异性靶向猪kit基因的sgrna,所述sgrna的核苷酸序列如seqidno.1、seqidno.2、seqidno.3、seqidno.4中的任一项所示。
本发明还提供了一种猪kit基因的cas9/sgrna共表达载体,其包括荧光素酶报告基因载体以及连接在所述荧光素酶报告基因载体上的靶向猪kit基因的sgrna,所述靶向猪kit基因的sgrna的核苷酸序列如seqidno.1、seqidno.2、seqidno.3、seqidno.4中的任一项所示。
根据所述的cas9/sgrna共表达载体,其中所述荧光素酶报告基因载体优选为px458载体。
本发明还提供了一种宿主细胞,其包括根据本发明所述的cas9/sgrna共表达载体。
本发明的方法可通过将sgrna筛选、流式细胞仪单细胞分选和qpcr检测拷贝数变化三者结合起来,可以在短时间内获得具有基因拷贝数变化的阳性单细胞克隆,极大地提高纠正基因结构突变的工作效率。
与传统纠正基因结构突变方法相比,本发明具有以下优点:利用crispr/cas9系统进行基因打靶,打靶效率较高;通过筛选活性较高的sgrna即可靶向各基因拷贝相同靶位点,实现拷贝数高效率删除;使用egfp基因作为流式分选的荧光报告基因,可以富集阳性细胞,进一步提高获得基因拷贝数变化的单细胞克隆的概率;通过qpcr检测拷贝数变化,不需要复杂的实验流程及仪器支持,适合在具有基本分子生物学设备的实验室进行大规模推广;适用范围广泛,适用于具有多个拷贝数的基因,不受特定细胞系限制。
与传统的策略不同,本发明为相同基因拷贝数的删除,在sgrna的设计上具有一定优势,即通过筛选一条活性较高的sgrna即可靶向各基因拷贝相同靶位点,进而通过nhej修复方式直接连接,实现拷贝数删除。利用crispr/cas9系统有效删除携带剪接突变位点的kit拷贝后,通过体细胞克隆可获得纠正kit基因结构突变的大白猪,恢复其正常的造血功能和免疫功能。
附图说明
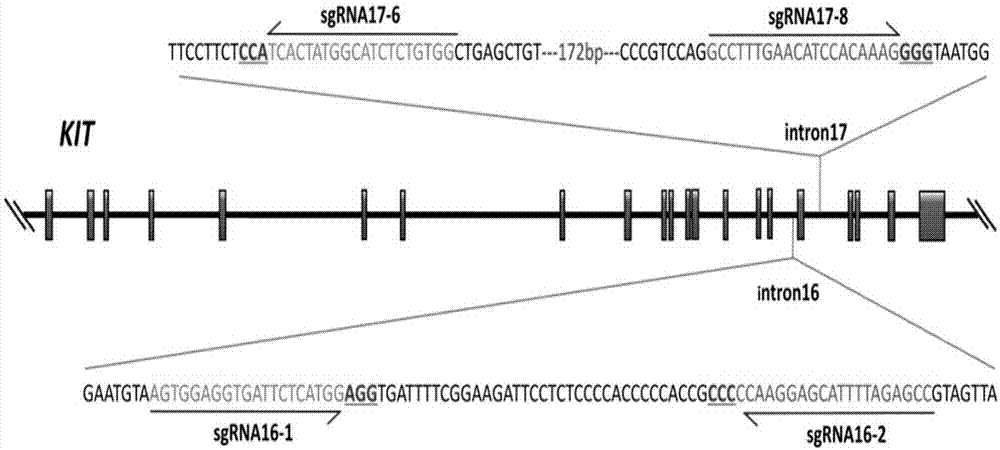
图1为sgrna在猪kit基因上的位置示意图。
图2为crispr/cas9系统对kit基因打靶效率的鉴定结果。
图3为t-a克隆测序检测打靶效率结果。
图4为qpcr鉴定单细胞克隆中kit基因拷贝数变化。
图5为kit基因编辑猪的拷贝数鉴定以及内含子17的g>a检测结果。
具体实施方式
以下结合具体实施例,对本发明作进一步说明。应理解,以下实施例仅用于说明本发明,而非用于限制本发明的范围。
下述实施例中所使用的试验方法如无特殊说明,均为常规方法。
所使用的材料、试剂等,如无特殊说明,为可从商业途径得到的试剂和材料。
实施例1
利用crispr/cas9技术纠正猪kit基因结构突变
一、高活性sgrna筛选
1、crispr/cas9打靶载体的构建
选定待编辑的目的基因(kit基因),选择sgrna打靶潜在目的区域序列,使用crisprdesign(http://crispr.mit.edu/)软件设计sgrna序列,选择评分较高的sgrna作为候选sgrna。
按照表1设计的sgrna,将合成的每对sgrna单链寡核苷酸变性退火成双链寡核苷酸片段,并在片段两侧加上磷酸基团,以便后续载体连接。
变性退火及磷酸化后将产物按照1:200比例加ddh2o稀释,以备后续酶切连接使用。
设计用于编辑猪kit基因的sgrna序列如下表1所示:
表1
将每条变性退火及磷酸化的sgrna双链寡核苷酸片段通过酶切和连接插入到px458空载体中,然后进行质粒化学转化、涂板、挑单克隆摇菌并测序,挑选测序正确的crispr/cas9打靶载体进行后续实验。
所设计的sgrna在猪kit基因上的位置如图1所示。
2、crispr/cas9打靶载体的筛选
1)猪肾细胞电转染
采用电转的方法将构建好的crispr/cas9系统转染1*106猪胎儿肾细胞细胞。
电转严格按照试剂盒和电转仪说明书操作。
2)流式分选egfp(增强型绿色荧光蛋白)阳性细胞
将构建好的crispr/cas9载体转染进细胞后,表达egfp绿色荧光,通过流式进行阳性筛选,分选出绿色荧光细胞(包含荧光报告基因信号),即为携带crispr/cas9载体的细胞。
3)t7e1酶切实验
实验均采用基因组提取试剂盒抽提细胞dna样品,参照kit基因登录号(cu929000.2),使用primerprimer5设计t7e1引物。
设计用于扩增删除区域的引物对如下表2所示:
表2
将上述得到的打靶细胞基因组dna作为模板,用上述表2设计的t7e1引物组成的引物对进行pcr扩增。将pcr产物胶回收或者纯化后进行变性退火,在变性退火后的pcr产物中加入0.5μlt7e1进行t7e1酶切实验,10%聚丙烯酰胺凝胶电泳分离鉴定。
4)t-a克隆测序鉴定高活性sgrna打靶效率
通过t7e1酶切实验检测阳性细胞打靶效率,采用imagej软件分析酶切结果,计算公式为突变
t-a克隆测序检测打靶效率,测序结果表明,在阳性细胞系中,sgrna16-1、sgrna16-2、sgrna17-6、sgrna17-8分选前(后)的突变效率分别为35.3%(88.9%)、27.5%(83.3%)、36.8%(50.0%)、15.0%(44.4%)(见图3)。
由结果可见,本发明设计的sgrna具有较高的打靶效率,可以用于kit基因拷贝数的删除。
二、单细胞克隆水平鉴定kit拷贝数变化
1、单细胞克隆培养
细胞转染48小时后(确保细胞状态良好),进行流式分选;分选前准备96孔板若干个,每个96孔板每孔加入150μl预热的条件培养基(50%新鲜全dmem和50%已使用全dmem混合过滤);分选后,放入细胞培养箱,三天后,每孔加入50μl全dmem培养基,一周后显微镜下观察细胞单克隆情况,并作相应标记,更换培养基。细胞单克隆堆积生长状态下,需要进行胰酶消化,加入培养基继续培养,细胞单克隆扩大培养至6孔板后,一部分提取基因组进行拷贝数鉴定,一部分冻存保种。
2、qpcr检测单细胞克隆拷贝数变化
如图4所示,qpcr结果显示,sgrna16-1介导的kit拷贝数删除单克隆样品中,编号1、4、5、9、18样品kit存在3个拷贝,其余样品kit为野生型,存在4个拷贝,单克隆样品阳性率为21.7%(5/23),排除每个样品本底水平两个kit拷贝,样品kit删除为10.9%(5/46);sgrna17-6介导的kit拷贝数删除样品中,样品3、7、15存在两个拷贝,样品11存在3个拷贝,单克隆样品阳性率为16.7%(4/24),排除每个样品本底水平两个kit拷贝,单克隆kit删除效率为14.6%(7/48)(图3)。
设计用于检测拷贝数变化的qpcr引物对如下表3所示:
表3
实施例2
利用体细胞核移植技术构建已纠正猪kit基因结构突变的编辑猪
1、体细胞核移植获得已纠正猪kit基因结构突变的编辑猪
从健康大白母猪体内采取挑选发育阶段适宜的卵巢,用注射器抽取卵巢表面直径在3-5mm的卵泡中的内含物,将内含物在tl-pva中稀释并重悬形成悬浊液。将悬浊液在37℃环境下静置至卵母细胞沉淀完全,将沉淀吸出置于体视镜下,用移液器或口吸管挑选卵周细胞完整的卵母细胞。将挑选的健康卵母细胞放入含有10%卵泡液、fsh、lh、egf的tcm-199中培养22h。再用移液器或口吸管将卵母细胞移到含有10%卵泡液、egf的tcm-199中继续培养22h。经过44h培养成熟后挑选已经排出第二极体的健康成熟卵母细胞作克隆胚胎用。
将上述制备的大白猪的含有已纠正猪kit基因结构突变的单细胞克隆,于5%co2、37℃饱和湿度的细胞培养箱培养,待细胞长至对数生长期时,即可用于核移植操作。
待卵母细胞体外培养成熟后,采用电融合法将含有已纠正猪kit基因结构突变的单细胞克隆进行体细胞核移植,并在24h之内进行胚胎移植,体细胞核移植及生产统计如表4所示。
表4
2、kit基因编辑猪的拷贝数鉴定以及内含子17的g>a检测
采取少量kit基因编辑猪的耳组织样提取基因组作为模板,通过qpcr进行拷贝数鉴定,并克隆测序,鉴定克隆猪kit基因内含子17的g>a突变情况。qpcr结果表明,在原有大白猪kit基因拷贝数的基础上,成功地实现了kit基因部分拷贝数的删除,且t-a克隆测序结果表明,位于内含子17的剪接突变位点被成功删除(见图5)。
序列表
<110>中山大学
<120>一种利用crisprcas9技术纠正猪kit基因结构突变的方法
<160>12
<170>siposequencelisting1.0
<210>1
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>1
agtggaggtgattctcatgg20
<210>2
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>2
ggctctaaaatgctccttgg20
<210>3
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>3
ccacagagatgccatagtga20
<210>4
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>4
gcctttgaacatccacaaag20
<210>5
<211>24
<212>dna
<213>人工序列(artificialsequence)
<400>5
caggctcatacatagaacgagatg24
<210>6
<211>19
<212>dna
<213>人工序列(artificialsequence)
<400>6
caggcacaggcttcactga19
<210>7
<211>23
<212>dna
<213>人工序列(artificialsequence)
<400>7
ttagtgatggctgtctgagatga23
<210>8
<211>25
<212>dna
<213>人工序列(artificialsequence)
<400>8
aggaggcaggtgaccgtattattac25
<210>9
<211>23
<212>dna
<213>人工序列(artificialsequence)
<400>9
gtcagtgctggcgatgagattag23
<210>10
<211>22
<212>dna
<213>人工序列(artificialsequence)
<400>10
acccagggtctcaaaagtccat22
<210>11
<211>22
<212>dna
<213>人工序列(artificialsequence)
<400>11
aagcttcaaacaggggtacaat22
<210>12
<211>23
<212>dna
<213>人工序列(artificialsequence)
<400>12
ccacttggaatgttaccctaatg23
- 还没有人留言评论。精彩留言会获得点赞!