Baeyer-Villiger单加氧酶、突变体及其在制备中长链二元羧酸中的应用的制作方法
本发明属于生物工程技术领域,具体涉及一种来源于铜绿假单胞菌(Pseudomonas aeruginosa)的Baeyer-Villiger单加氧酶及其突变体、含有该单加氧酶基因的重组表达载体和重组表达转化体,以及利用该重组Baeyer-Villiger单加氧酶催化长链酮酸或长链酮酯的Baeyer-Villiger氧化反应,生成长链酯酸或二酯,进而制备α,ω-二元羧酸的方法。
背景技术:
α,ω-二元羧酸因其官能团的特殊性,可以作为单体合成高聚物,如聚酯类、聚酰胺类等各种功能塑料,也可用来合成润滑剂、增塑剂、涂料、高级香料、缓蚀剂等各类精细化学品。化学法合成中长链二元羧酸需要以短链二元羧酸为底物,经过多步反应延伸碳链制得,存在着操作繁琐、得率低等问题,而且化学法合成的都是饱和二元羧酸,链长有限,碳原子数一般不超过13,不饱和长链二元羧酸的化学法合成还未取得突破。相较于化学方法,生物酶法转化合成α,ω-二元羧酸具有专一性好、选择性高、催化反应条件(温度、pH等)温和、能耗低、污染小等特点,受到人们的青睐。尤其是以种类繁多、来源丰富的植物油为底物经生物转化生成α,ω-二元羧酸,符合可持续发展理念,具有广阔的发展前景。
在目前的研究中,由生物法制备α,ω-二元羧酸主要包括两条路径:一条途径是将热带假丝酵母中的脂肪酸β-氧化途径切断,利用P450酶系催化脂肪酸的ω-氧化从而合成二元羧酸;另一条途径需要水合酶、醇脱氢酶、Baeyer-Villiger单加氧酶等多酶级联催化,将不饱和脂肪酸转化生成酯酸,进一步水解生成二元羧酸。在后一条路径中,Baeyer-Villiger单加氧酶(缩写为BVMO)催化酮酸化合物的不对称Baeyer-Villiger氧化生成相应的酯酸,是该途径中的关键酶。
根据催化底物的特异性,可以将BVMO分为环己酮单加氧酶(CHMO)、环戊酮单加氧酶(CPMO)、4-羟基苯乙酮单加氧酶(HAPMO)和直链脂肪酮单加氧酶(AKMO)等不同类别。其中,直链脂肪酮单加氧酶也可以催化酮酸化合物的氧化,但目前对于这类酶的报道较少。Kirschner等对来自荧光假单胞菌的PfBVMO进行了克隆表达,该酶对碳原子数为8-12的脂肪酮活性较高(Appl.Microbiol.Biotechnol.,2007,73:1065-1072)。Rehdorf等克隆得到来自恶臭假单胞菌的PpBVMO,该酶对芳香酮、环酮、脂肪酮(3-癸酮、2-十一酮、2-十二酮等)都有较高的活性(Biotechnol.Lett.,2007,29:1393-1398)。Altenbuchner等克隆了来自铜绿假单胞菌的单加氧酶MEK700,该酶的底物谱较广,对直链酮、芳香酮和环酮类化合物都有活性(Appl.Microbiol.Biotechnol.,2008,77:1251-1260)。Bisagni等克隆得到迪茨氏菌属中的BVMO,该酶偏好的催化底物是直链脂肪酮,包括2-壬酮、2-庚酮、3-癸酮等(J.Mol.Catal.B:Enzym.,2014,109:161-169)。
Song等在2013年构建了利用AKMO参与植物来源脂肪酸的生物转化,生成中链二元羧酸的技术路径(Angew.Chem.Int.Ed.,2013,52:2534-2537)。在以10-羰基硬脂酸为底物进行催化反应时,因用到的两种BVMO(恶臭假单胞菌中的PpBVMO和荧光假单胞菌中的PfBVMO)对氧插入位置的偏好性不同,故分别转化生成两种酯。其中,PpBVMO偏好在羰基靠近羧基的一侧插入氧,生成的酯称为“常规酯”,而PfBVMO偏好在远离羧基的一侧插入氧,生成的酯称为“非常规酯”。在底物上载量为1mM时,PpBVMO参与的反应路径的主要水解产物为9-羟基壬酸和壬酸,分析得率高于60%;而PfBVMO参与的反应路径的主要水解产物为辛醇和癸二酸,分析得率也高于60%。
虽然PfBVMO能够催化长链酮酸氧化生成非常规酯,进而水解获得目标产物α,ω-二元羧酸(癸二酸),但该酶区域选择性不高,非常规酯与常规酯的比率仅为71:29,因此底物利用率较低,且难以获得结构单一的α,ω-二元羧酸,产品分离纯化的成本高。而常规酯水解产物α,ω-羟基酸需要再经过多步的化学法或酶法氧化才能生成α,ω-二元羧酸,反应步骤多,底物利用率低,并且存在环境污染问题(Adv.Synth.Catal.,2014,356:1782–1788;Adv.Synth.Catal.,2016,358:3084-3092)。
技术实现要素:
本发明针对目前已报道的BVMO催化长链酮酸的Baeyer-Villiger氧化反应的区域选择性差,获得的非常规酯的比率低,合成α,ω-二元羧酸步骤繁琐的问题,提供一种“非常规”区域选择性高的Baeyer-Villiger单加氧酶PaBVMO,利用该酶分别催化氧化10-羰基十八烷酸和9-羰基十八烷酸得到癸二酸一辛酯和壬二酸一壬酯,经过化学水解得到癸二酸和壬二酸,可以显著缩短其合成路径。
本发明的目的可以通过以下技术方案来实现:
本发明第一方面:提供一种“非常规”区域选择性高的Baeyer-Villiger单加氧酶。
一种“非常规”区域选择性高的Baeyer-Villiger单加氧酶,其是如下(a)或(b)的蛋白质:
蛋白质(a):由SEQ ID No.2所示氨基酸序列组成的蛋白质;
蛋白质(b):SEQ ID No.2所示氨基酸序列中经过取代、缺失或添加几个氨基酸且具有Baeyer-Villiger单加氧酶活性的由(a)衍生的蛋白质。
所述蛋白质(a)通过基因组挖掘的方法获得,所设计的挖掘方法具体为,以来自荧光假单胞菌Pseudomonas fluorescens的Baeyer-Villiger单加氧酶PfBVMO的氨基酸序列作为参考序列,在NCBI数据库中进行pBLAST搜索,选择一批功能预测为Baeyer-Villiger单加氧酶的氨基酸序列,其与PfBVMO的氨基酸序列一致性为30-70%,对这些蛋白进行克隆,构建重组大肠杆菌细胞,测定表达的重组蛋白的10-羰基硬脂酸的Baeyer-Villiger单加氧反应活性,以及区域选择性等参数,对所克隆的酶进行综合比较和筛选,最终获得区域选择性最高、来源于铜绿假单胞菌(Pseudomonas aeruginosa)的Baeyer-Villiger单加氧酶PaBVMO。该酶是依据登录号为WP_003087250.1,预测为Baeyer-Villiger单加氧酶的蛋白质的核酸序列设计引物,从保藏号为CGMCC 1.9047的铜绿假单胞菌中克隆获得。其中保藏号为CGMCC 1.9047的铜绿假单胞菌来自中国普通微生物菌种保藏管理中心。
所述蛋白质(b)是在蛋白质(a)的基础上经过一个或多个氨基酸的替换、缺失或添加得到的具有Baeyer-Villiger单加氧酶活性的衍生蛋白质。在通过筛选获得高区域选择性的酶PaBVMO的基础上,对野生型PaBVMO进行蛋白质工程改造,以进一步提高该酶的活性。通过易错PCR技术及对PaBVMO的活性口袋和底物通道附近的氨基酸残基进行半理性设计,发现在SEQ ID No.2所示氨基酸序列的基础上,对第56位天冬酰胺残基、第141位的丝氨酸残基、第257位苯丙氨酸残基、第310位的丙氨酸残基、第313位苏氨酸残基分别或共同进行氨基酸残基替换,仍具有Baeyer-Villiger单加氧酶活性。在此基础上,通过精心设计、筛选获得了酶活性显著提高的突变体。
优选的,所述Baeyer-Villiger单加氧酶突变体,为野生Baeyer-Villiger单加氧酶PaBVMO的氨基酸残基发生如下任意一种情况的突变获得的突变体:
(1)野生型Baeyer-Villiger单加氧酶PaBVMO的氨基酸序列中的第56位天冬酰胺替换为亮氨酸,命名为PaBVMON56L;
(2)野生型Baeyer-Villiger单加氧酶PaBVMO的氨基酸序列中的第141位丝氨酸替换为亮氨酸,命名为PaBVMOS141L;
(3)野生型Baeyer-Villiger单加氧酶PaBVMO的氨基酸序列中的第257位苯丙氨酸替换为谷氨酰胺,命名为PaBVMOF257Q;
(4)野生型Baeyer-Villiger单加氧酶PaBVMO的氨基酸序列中的第310位丙氨酸替换为半胱氨酸,命名为PaBVMOA310C;
(5)野生型Baeyer-Villiger单加氧酶PaBVMO的氨基酸序列中的第313位苏氨酸替换为缬氨酸,命名为PaBVMOT313V;
(6)野生型Baeyer-Villiger单加氧酶PaBVMO的氨基酸序列中第141位丝氨酸替换为亮氨酸,且第257位苯丙氨酸替换为谷氨酰胺,且第310位丙氨酸替换为半胱氨酸,命名为PaBVMOS141L/F257Q/A310C;
(7)野生型Baeyer-Villiger单加氧酶PaBVMO的氨基酸序列中第56位天冬酰胺替换为亮氨酸,且第141位丝氨酸替换为亮氨酸,且第257位苯丙氨酸替换为谷氨酰胺,且第310位丙氨酸替换为半胱氨酸,且第313位苏氨酸替换为缬氨酸,命名为PaBVMON56L/S141L/F257Q/A310C/T313V。
所述突变体的获得方法具体为:以来源于铜绿假单胞菌(Pseudomonas aeruginosa)中的Baeyer-Villiger单加氧酶野生酶PaBVMO基因作为模板,利用随机突变引物进行易错PCR构建随机突变库,对所得到的突变库进行高通量筛选,得到活力提高的突变体PaBVMON56L、PaBVMOS141L、PaBVMOF257Q、PaBVMOA310C和PaBVMOT313V;然后以突变体PaBVMOS141L的基因为模板,利用含有突变点的突变引物(选取需要进行突变的氨基酸位点上下游各15-20bp的一段碱基序列,将突变位点的碱基替换为突变后氨基酸的密码子,作为PCR正向引物,其反向互补序列即为PCR反向引物),通过PCR方法扩增,得到突变体基因,表达获得活性更高的PaBVMOS141L/F257Q/A310C和PaBVMON56L/S141L/F257Q/A310C/T313V。
其中所述易错PCR扩增为本领域常规技术,可选的PCR反应的体系(50μL)为:模板0.5-20ng,10×rTag buffer 5μL,dNTP(各2.0mM)5μL,MgSO4(25mM)2μL,MnCl2(100μM)5μL,一对突变引物(20μM)各1μL,1个单位的rTaq酶,加超纯水至50μL。
可选的所述易错PCR扩增的程序为:(1)94℃变性3min;(2)94℃变性10s,(3)60℃退火30s,(4)68℃延伸90s,步骤(2)-(4)共进行30个循环,最后72℃延伸10min,4℃保藏产物。
其中所述定点突变PCR扩增为本领域常规技术,可选的PCR反应的体系(50μL)为:模板5-20ng,10×KOD plus buffer 5μL,dNTP(各2.0mM)5μL,MgSO4(25mM)2μL,一对突变引物(20μM)各1μL,1个单位的KOD酶,加超纯水至50μL。
可选的所述定点突变PCR扩增的程序为:(1)94℃变性5min;(2)94℃变性30s,(3)60℃退火1min,(4)68℃延伸90s,步骤(2)-(4)共进行30个循环,最后68℃延伸10min,4℃保藏产物。
本发明中提及的区域选择性是指Baeyer-Villiger单加氧酶催化长链酮酸氧化生成的非常规酯和常规酯的摩尔比。其中,氧插入在酮酸中酮基的非羧基取代的一侧生成的酯称为非常规酯;反之,氧插入酮酸中酮基的羧基取代的一侧生成的酯为常规酯。
本发明所述的PaBVMO的编码基因来源于保藏号为CGMCC 1.9047的铜绿假单胞菌Pseudomonas aeruginosa。其编码DNA的具体制备方法包括:以铜绿假单胞菌的基因组DNA为模板,采用本领域常规技术方法(如聚合酶链式反应,PCR),获得编码所述单加氧酶PaBVMO的完整DNA序列。其中设计的合成引物,如SEQ ID No.3(上游引物)和SEQ ID No.4(下游引物)所示:上游引物:5’-CCGGAATTCATGAGTACCCAACCCACC-3’,其中下划线所示序列为限制性内切酶EcoR I的酶切位点;下游引物:5’-CCCAAGCTTTCATGCGGGTACCCCTTC-3’,其中下划线所示序列为限制性内切酶Hind III的酶切位点。
本发明中所述单加氧酶PaBVMO全长基因的核苷酸序列如SEQ ID No.1所示,全长为1545个核苷酸碱基。其编码序列(CDS)从第1个碱基起至第1542个碱基止,起始密码子为ATG,终止密码子为TGA,无内含子,该基因编码的蛋白质的氨基酸序列如序列表中SEQ ID No.2所示。
由于密码子的简并性,编码SEQ ID No.2的氨基酸序列的碱基序列不仅仅局限于SEQ ID No.1。另外,还可以通过适当引入替换、缺失、改变、插入或增加来提供一个多聚核苷酸的同系物。本发明中多聚核苷酸的同系物可以通过对碱基序列SEQ ID No.1的一个或多个碱基在保持酶活性范围内进行替换、缺失或增加来制得。
SEQ ID No.1的同系物也指启动子变体。在所述的碱基序列之前的启动子或信号序列可通过一个或多个核苷酸的替换、插入或缺失而改变,但这些改变对启动子的功能没有负面影响。而且通过改变启动子的序列或甚至用来自不同种生物体的更有效的启动子进行替换,可提高目标蛋白的表达水平。
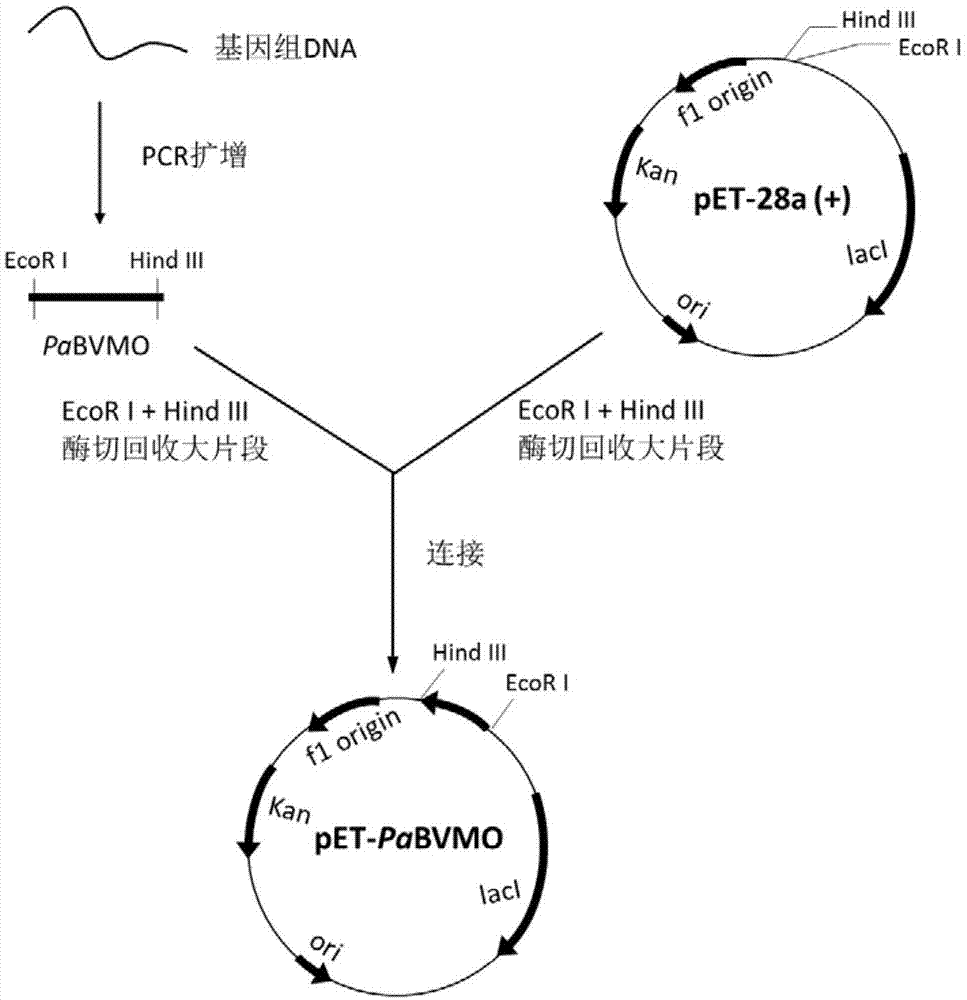
本发明还提供了含有上述单加氧酶基因核酸序列的重组表达载体。所述重组表达载体可通过本领域常规方法,将上述单加氧酶基因序列DNA片段连接到各种表达载体上构建而成。所述的表达载体较佳的包括本领域常规的各种质粒载体,优选的为pET28a质粒。较佳的,作为示例,可通过下述方法制得本发明所述的重组表达载体:将通过PCR扩增所得的单加氧酶PaBVMO的基因序列DNA片段用限制性内切酶EcoR I和Hind III双酶切,同时将空载质粒pET28a用限制性内切酶EcoR I和Hind III双酶切,回收上述酶切后的单加氧酶PaBVMO的基因DNA片段以及pET28a质粒,利用T4DNA连接酶连接,构建获得包含所述单加氧酶PaBVMO基因的重组表达载体pET28a-PaBVMO。
本发明还提供了包含上述单加氧酶重组表达载体的重组表达转化体。所述重组表达转化体可通过将上述重组表达载体转化至宿主细胞中制得。所述宿主细胞为本领域常规的宿主细胞,只要能满足重组表达载体可稳定地自行复制,并且其所携带的单加氧酶基因可被有效表达即可。所述宿主细胞优选为大肠杆菌,更优选的为:大肠杆菌E.coli BL21(DE3)。将所述重组表达载体转化至大肠杆菌E.coli BL21(DE3)中,即可获得本发明优选的基因工程菌株。例如,将重组表达载体pET28a-PaBVMO转化至大肠杆菌E.coli BL21(DE3)中,获得重组大肠杆菌E.coli BL21(DE3)/pET28a-PaBVMO。
本发明还提供了上述重组单加氧酶的制备方法。所述重组单加氧酶的制备方法较佳地为:培养如上所述的重组表达转化体,分离获得重组表达的单加氧酶。其中所述重组表达转化体培养所用的培养基为本领域任何可使转化体生长并产生本发明的重组单加氧酶的培养基。所述培养基优选LB培养基,其配方为:蛋白胨10g/L,酵母膏5g/L,NaCl 10g/L,pH 7.0。培养方法和培养条件没有特殊的限制,可以根据宿主细胞类型和培养方法等因素的不同,按本领域常规知识进行适当的选择,只要使转化体能够生长并生产所述单加氧酶即可。重组表达转化体培养的具体操作可按本领域常规操作进行。优选的,将本发明所述的重组大肠杆菌,例如E.coli BL21(DE3)/pET28a-PaBVMO,接种至含卡那霉素的LB培养基中,37℃培养,当培养液的光密度OD600达到0.5-1.0(优选0.6)时,加入终浓度为0.1-1.0mmol/L(优选0.5mmol/L)的异丙基-β-D-硫代半乳糖苷(IPTG)进行产酶诱导,在16℃继续培养24h,即可高效表达本发明所述的单加氧酶PaBVMO。培养结束后,离心收集沉淀的菌体细胞,即为重组表达转化体的静息细胞;将收获的细胞悬浮于甘氨酸-NaOH缓冲液(100mM,pH 9.0)中,超声破碎,破碎液离心,收集上清液,即可获得所述重组单加氧酶PaBVMO的粗酶液;将离心收获的细胞沉淀冷冻干燥,可以获得冻干细胞,有利于长期存放,方便以后使用。
单加氧酶PaBVMO的活性测定:将含0.2mmol/L 10-羰基十八烷酸和0.1mmol/L NADPH的1mL反应体系(100mmol/L磷酸钾缓冲液,pH 9.0)预热至25℃,然后加入适量的单加氧酶PaBVMO,混合均匀,25℃保温反应,在分光光度计上检测340nm处NADPH的吸光度变化,记录一定时间内吸光度的变化值。
根据下式计算得到酶活力:
酶活力(U)=EW×V×103/(6220×l)
式中,EW为1分钟内340nm处吸光度的变化;V为反应液的体积,单位为mL;6220为NADPH的摩尔消光系数,单位为L/(mol·cm);l为光程距离,单位为cm。1个酶活力单位(U)对应于上述条件下每分钟氧化1μmol NADPH所需的酶量。
本发明还提供了所述Baeyer-Villiger单加氧酶在催化长链酮酸或长链酮酯类化合物转化,制备α,ω-二元羧酸中的应用。其中所述长链酮酸或长链酮酯类化合物的化学结构如下式1或2所示,式1为长链酮酸,式2为长链酮酯。
其中,式1中,m=5-9,n=7-8;
式2中,m=5-9,n=7-8,R为-CH3或者-C2H5。
所述长链酮酸或长链酮酯的Baeyer-Villiger单加氧反应,可按下述示例性方法进行:在pH 7.5-9.5的甘氨酸-氢氧化钠缓冲液中,在葡萄糖脱氢酶、葡萄糖和NADP+的存在下,在所述单加氧酶PaBVMO的作用下,催化所述长链酮酸或长链酮酯的Baeyer-Villiger单加氧反应。反应结束后,采用化学法对生成的脂肪酸酯进行水解,获得相应的α,ω-二元酸。所述应用中,底物在反应液中的浓度可以为0.1-100mmol/L。根据所采用的反应体系,所述单加氧酶的用量可以为1-500U/L。酶促长链酮酸或长链酮酯的Baeyer-Villiger单加氧反应时,辅酶NADPH氧化生成NADP+,为了进行辅酶NADPH的循环再生,向反应体系中额外添加葡萄糖和来自巨大芽孢杆菌的葡萄糖脱氢酶(中国现代医学杂志,2007,17:172-174)。取决于不同的反应体系,葡萄糖脱氢酶的活力单位上载可以与所述Baeyer-Villiger单加氧酶相等。葡萄糖与底物的摩尔比可以为1.0-1.5,额外添加的NADP+的用量可以为0-1.0mmol/L。所述缓冲液可以是本领域常规的任何缓冲液,比如柠檬酸钠、磷酸钠、磷酸钾、Tris-HCl或者甘氨酸-氢氧化钠缓冲液,只要其pH范围在5.0-10.0即可,优选pH范围为7.5-9.5,更优选pH 9.0。所述缓冲液的浓度可以为0.05-0.2mol/L。所述的酶促不对称还原反应的温度可以是15-35℃,优选25℃。反应过程中,间歇取样测定反应转化率,反应时间以底物完全转化或反应转化率停止增长的时间为准,一般为1-24小时。反应转化率和区域选择性可采用气相色谱法进行分析。本发明中分别通过分析常规酯和非常规酯的水解产物ω-羟基酸和α,ω-二元酸的浓度进行定量和区域选择性计算。优选的,使用HP-5MS毛细管色谱柱(30m length,0.25μm film thickness,Agilent Technologies)进行分析,载气为氮气,检测器为氢火焰离子化检测器(FID)。进样口温度为280℃,检测器温度为280℃。
酶促区域选择性单加氧反应结束后,采用常规方法制备获得目标产物二元羧酸。优选的,用有机溶剂对反应产物进行萃取,挥发除去溶剂,然后加入碱溶液(NaOH或KOH),加热到60-80℃对反应生成的脂肪酸酯产物进行水解,生成二元羧酸盐。然后在水解液中加入强酸,获得二元羧酸的沉淀,过滤得到目标产物二元羧酸。
在符合本领域常识的基础上,上述各优选条件,可任意组合,即得到本发明较佳实施例。
本发明所用的原料或试剂除特别说明之外,均市售可得。
附图说明
图1为重组表达质粒pET-PaBVMO的构建示意图。
具体实施方式
下面通过实施例进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
下列实施例中的材料来源为:
表达质粒pET28a购自上海Novagen公司。
大肠杆菌E.coli DH5α和E.coli BL21(DE3)感受态细胞,2×Taq PCR MasterMix,琼脂糖凝胶DNA回收试剂盒购自北京天根生化科技有限公司。
实施例1“非常规”区域选择性Baeyer-Villiger单加氧酶的筛选
以具有较高“非常规”区域选择性的Baeyer-Villiger单加氧酶PfBVMO为探针(序列登录号AAC36351.2),在NCBI中通过蛋白序列比对,选取一批与探针序列相似度在20%-70%的序列,设计引物,进行基因克隆。将PCR扩增得到的目的片段经酶切、连接接到质粒pET28a上,转化至E.coli DH5α中,经菌落PCR验证后送金斯瑞生物科技有限公司进行序列测定。对测序结果与目的序列一致的菌提取重组质粒,将质粒再一次转化入E.coli BL21(DE3)中,进行目的酶的过量表达和区域选择性筛选。
以1mM 10-羰基十八烷酸为底物,用重组表达整细胞进行1mL反应。反应体系如下:1mL Tris-HCl缓冲液(100mM,pH 7.5),1mM 10-羰基十八烷酸,1g/L Tween-80,5mM葡萄糖,0.2mM NADP+,50g/L重组大肠杆菌湿细胞,1g/L BmGDH冻干酶粉,于30℃震荡反应8h。反应结束,加入20%H2SO4调节pH至2以下,终止反应。加入等体积乙酸乙酯及NaCl至饱和后振荡萃取,离心3min。吸取上层乙酸乙酯相,挥发除去溶剂,加入40μL的KOH溶液(1M)溶解,60℃水解2h。反应结束,加入NaCl至饱和,加入160μL 20%H2SO4、400μL乙酸乙酯(含0.5mM正十二烷和0.5mM软脂酸为内标)震荡萃取。吸取上层乙酸乙酯相,加入一定量无水硫酸钠进行干燥。以氮气为载气,用HP-5MS气相色谱柱进行气相色谱分析水解产物,通过检测正辛醇(非常规酯水解产物)和9-羟基壬酸(常规酯水解产物)的比例,表征克隆的单加氧酶的区域选择性。其中,来源于铜绿假单胞菌(Pseudomonas aeruginosa)的Baeyer-Villiger单加氧酶PaBVMO具有最高的区域选择性,为89:11。
实施例2单加氧酶PaBVMO基因的克隆及重组表达转化体构建
根据铜绿假单胞菌(Pseudomonas aeruginosa)中Genebank收录的预测为Baeyer-Villiger单加氧酶的基因序列(Genebank登录号:WP_003087250.1)为依据,设计PCR引物如下:
上游引物:CCGGAATTCATGAGTACCCAACCCACC;
下游引物:CCCAAGCTTTCATGCGGGTACCCCTTC。
其中,上游引物的下划线部分为EcoR I的酶切位点,下游引物的下划线部分为Hind III的酶切位点。
以保藏号为CGMCC1.9047的铜绿假单胞菌(Pseudomonas aeruginosa)的基因组DNA为模板,进行PCR扩增。PCR扩增体系的总体积为50μL,具体为:2x Taq PCR MasterMix 25μL,上游引物和下游引物各1.5μL(0.4μmol/L),DNA模板2μL(1μg)和ddH2O 20μL。PCR扩增程序为:(1)95℃,预变性3min;(2)94℃,预变性30s;(3)55℃,退火30s;(4)72℃,延伸90s;步骤(2)-(4)重复30次;(5)72℃,延伸10min,冷却至4℃。PCR产物经琼脂糖凝胶电泳纯化,利用琼脂糖凝胶DNA回收试剂盒回收1500bp左右目的条带,30℃,用限制性内切酶EcoR I和Hind III双酶切,与同时使用限制性内切酶EcoR I和Hind III双酶切的空载质粒pET28a,利用T4DNA连接酶连接,构建获得包含所述单加氧酶PaBVMO基因的重组表达载体pET28a-PaBVMO(见图1),转化到大肠杆菌E.coli DH5α感受态细胞中,在含有卡那霉素的抗性平板上对阳性重组体进行筛选,挑取克隆体,菌落PCR验证阳性克隆。培养重组菌,待质粒扩增后提取质粒,重新转化至大肠杆菌E.coli BL21(DE3)感受态细胞中,转化液涂布到含有卡那霉素的LB平板上,37℃倒置培养过夜,获得阳性大肠杆菌重组表达转化体E.coli/pET28a-PaBVMO,菌落PCR验证阳性克隆。抽提质粒,送金斯瑞生物科技有限公司进行序列测定,其基因编码序列如序列表SEQ ID No.1所示。
实施例3单加氧酶PaBVMO的随机突变
利用易错PCR技术向PaBVMO基因引入随机核苷酸突变,其中所用引物如下:
上游引物:CCGGAATTCATGAGTACCCAACCCACC;
下游引物:CCCAAGCTTTCATGCGGGTACCCCTTC。
其中,模板为如实施例2获得的PaBVMO基因重组质粒。
PCR反应的体系(50μL)为:模板0.5-20ng,10×rTag buffer 5μL,dNTP(各2.0mM)5μL,MgSO4(25mM)2μL,MnCl2(100μM)5μL,一对突变引物(20μM)各1μL,1个单位的rTaq酶,加超纯水至50μL。
PCR扩增的程序为:(1)94℃变性3min;(2)94℃变性10s,(3)60℃退火30s,(4)68℃延伸90s,步骤(2)-(4)共进行30个循环,最后72℃延伸10min,4℃保藏产物。
PCR扩增产物纯化之后,用EcoR I和Hind III分别对易错PCR扩增产物和载体pET28a进行双酶切,两者的回收产物,用T4连接酶16℃下连接5h,转化E.coli BL21(DE3)感受态细胞,并均匀涂布于含有50μg/mL卡那霉素的LB琼脂平板,37℃过夜培养,构建随机突变体文库,并进行筛选。筛选得到活性为母本活性2.1-3.5倍的突变体,经DNA测序发现其突变位点分别为SEQ ID No.2所示氨基酸序列中的第56位、第141位、第257位、第310位、第313位,突变情况是第56位天冬酰胺残基替换为亮氨酸残基,第141位丝氨酸残基替换为亮氨酸残基,第257位苯丙氨酸残基替换为谷氨酰胺残基,第310位丙氨酸残基替换为半胱氨酸残基,第313位苏氨酸残基替换为缬氨酸残基,所得突变体表示为PaBVMON56L、PaBVMOS141L、PaBVMOF257Q、PaBVMOA310C、PaBVMOT313V,简写表示为:N56L、S141L、F257Q、A310C和T313V。
实施例4单加氧酶PaBVMO的组合突变
定点突变采用QuikII Site-Directed Mutagenesis Kit(Stratagene,Catalog#200522)方案进行操作。首先设计含有突变点的突变引物如下:
突变F257Q:
上游引物:GGCTTCACCCAAGCCCCGCAGGTGATGAAGCTG,见SEQ ID No.5。
下游引物:CTGCGGGGCTTGGGTGAAGCCCAGCACCCGCCC,见SEQ ID No.6。
突变A310C:
上游引物:CTGGCTGCCTGCAATTCCACGGTGATCACCGAA,见SEQ ID No.7。
下游引物:CGTGGAATTGCAGGCAGCCAGGGCCGGATAGTA,见SEQ ID No.8。
突变N56L:
上游引物:AGAGTCAACCTTTACCCTGGCTGCGCCTGCGAC,见SEQ ID No.9。
下游引物:GCCAGGGTAAAGGTTGACTCTCCAGGTGCCGCC,见SEQ ID No.10。
突变T313V:
上游引物:GCCAATTCCGTTGTGATCACCGAAGGCATCCGC,见SEQ ID No.11。
下游引物:GGTGATCACAACGGAATTGGCGGCAGCCAGGGC,见SEQ ID No.12。
以实施例3获得的突变体PaBVMOS141L质粒为模板,PCR反应的体系(50μL)为:模板5-20ng,10×KOD plus buffer 5μL,dNTP(各2.0mM)5μL,MgSO4(25mM)2μL,一对突变引物(20μM)各1μL,1个单位的KOD酶,加超纯水至50μL。
PCR扩增的程序为:(1)94℃变性5min;(2)94℃变性30s,(3)60℃退火1min,(4)68℃延伸90s,步骤(2)-(4)共进行30个循环,最后68℃延伸10min,4℃保藏产物。
PCR扩增产物在37℃经内切酶DpnI消化2h后,转化E.coli BL21(DE3)感受态细胞,并均匀涂布于含有50μg/mL卡那霉素的LB琼脂平板,37℃过夜培养,挑选单克隆送测序。将测序结果进行比对,确认突变前后基因序列及相应的氨基酸序列的差异。
其中,S141L/F257Q/A310C突变构建过程为:先以突变体S141L的全长基因序列为模板,用突变体F257Q的PCR引物构建突变体S141L/F257Q,再以此突变体为模板,用突变体A310C的PCR引物构建突变体S141L/F257Q/A310C。
其中,N56L/S141L/F257Q/A310C/T313V突变构建过程为:先以突变体S141L/F257Q/A310C的全长基因序列为模板,用突变体N56L的PCR引物构建N56L/S141L/F257Q/A310C,再以此突变体为模板,用突变体T313V的PCR引物构建突变体N56L/S141L/F257Q/A310C/T313V。
实施例5重组表达质粒的构建和重组表达转化体的制备
将实施例2-4构建、PCR扩增、回收得到的Baeyer-Villiger单加氧酶基因DNA片段在37℃用限制性内切酶EcoR I和Hind III进行双酶切12h,经琼脂糖凝胶电泳纯化,利用琼脂糖凝胶DNA回收试剂盒回收目的片段。将目的片段在T4DNA连接酶的作用下,与同样经过EcoR I和Hind III双酶切的质粒pET28a,在16℃下连接过夜得到重组质粒。
将上述重组表达质粒转化到大肠杆菌E.coli DH5α感受态细胞中,在含有卡那霉素的抗性平板上对阳性重组体进行筛选,挑取克隆体,菌落PCR验证阳性克隆。培养重组菌,待质粒扩增后提取质粒,重新转化至大肠杆菌E.coli BL21(DE3)感受态细胞中,转化液涂布到含有卡那霉素的LB平板上,37℃倒置培养过夜,获得阳性大肠杆菌重组表达转化体,菌落PCR验证阳性克隆。
实施例6重组单加氧酶的表达
在无菌环境中,将实施例5所得的重组大肠杆菌,接种至含卡那霉素(终浓度50μg/mL)的LB培养基(10g/L蛋白胨,5g/L酵母膏,10g/L NaCl,pH 7.0)中,37℃振荡培养过夜,按1%(v/v)的接种量接入装有100mL LB培养基的500mL摇瓶中,置37℃摇床振荡培养,当培养液的OD600达到0.6时,加入终浓度为0.2mmol/L的IPTG作为诱导剂,诱导目的蛋白表达,于16℃诱导培养24h后,将培养液离心,收集细胞,并用生理盐水洗涤两次,得到静息细胞。将获得的静息细胞用10倍体积的磷酸钠缓冲液(20mM,pH 7.0)重新悬浮,过筛后预冷到4℃,用高压匀浆机进行破碎,压力800bar。在4℃对破碎液进行离心,12000×g,20min,收集上清液,即为所述重组单加氧酶粗酶液。
使用分光光度计,通过检测340nm处吸光值变化的方式,测定单加氧酶的活力。测定方法如下:于1mL反应体系(100mmo1/L甘氨酸-氢氧化钠缓冲液,pH 9.0)中,加入0.2mmol/L 10-羰基十八烷酸,0.1mmol/L NADPH,25℃保温2分钟后加入适量粗酶液,迅速混匀,检测340nm处吸光值的变化,粗酶液中酶的比活见表1。粗酶液经聚丙烯酰胺凝胶电泳图分析,重组蛋白以部分可溶的形式存在。
以10-羰基十八烷酸为底物,利用重组表达整细胞进行1mL反应。通过气相色谱分析水解产物对单加氧酶的区域选择性进行表征,操作方法同实施例1。单加氧酶对10-羰基十八烷酸的区域选择性测定结果见表1
表1PaBVMO及其突变体对10-羰基十八烷酸的活性和区域选择性比较
酶活力的计算公式为:酶活力(U)=EW×V×103/(6220×l)。式中,EW为1min内340nm处吸光度的变化;V为反应液的体积,单位mL;6220为NADPH的摩尔消光系数,单位L/(mol·cm);l为光程距离,单位cm。Baeyer-Villiger单加氧酶的活力单位定义为,在上述条件下,每分钟催化1μmol NADPH氧化所需的酶量定义为一个活力单位。
将粗酶液倒入平底盘中,置于-80℃冰箱冷冻过夜后放入冻干机进行冷冻干燥,获得冻干酶粉,制备好的酶粉置于4℃储藏备用。
实施例7-12重组单加氧酶PaBVMON56L/S141L/F257Q/A310C/T313V催化长链酮酸的Baeyer-Villiger氧化
在0.4mL甘氨酸-氢氧化钠缓冲液(100mmol/L,pH 9.0)中加入5U如实施例6制备的PaBVMON56L/S141L/F257Q/A310C/T313V冻干酶粉和5U葡萄糖脱氢酶冻干酶粉,加入终浓度为1mmol/L的系列长链酮酸,再加入终浓度为0.1mmol/L的NADP+和5mmol/L的葡萄糖。在20℃,1000rpm振荡反应一定时间。反应结束后用20%H2SO4调节pH至2以下,终止反应。加入等体积乙酸乙酯振荡萃取,12000×g离心3min,吸取上层有机相,挥发除去溶剂,然后加入40μL浓度为1mol/L的KOH溶液,于60℃反应2h,使单加氧反应产物水解生成相应的醇、羟基酸或二酸,进而进行检测。反应结束,加入160μL 20%H2SO4和400μL乙酸乙酯(含0.5mM正十二烷和0.5mM软脂酸作为内标)振荡萃取。吸取上层有机相并加入一定量无水硫酸钠干燥过夜,水解产物9-羟基壬酸和癸酸需先经过硅烷衍生化再进行气相色谱分析。衍生化条件为:40μL水解反应萃取液,40μL吡啶和20μL N,O-双(三甲基硅烷基)三氟乙酰胺(BSTFA,含1%四甲基硅烷),于75℃保温25min。用气相色谱分析测定水解产物的浓度和PaBVMON56L/S141L/F257Q/A310C/T313V对长链酮酸的区域选择性,使用的色谱柱为HP-5MS。
水解产物以及相应的具体气相色谱分析条件如下:
实施例7-12的气相色谱分析中,载气为氮气,进样口温度为280℃,检测器温度为280℃,其他条件如下:
实施例7:底物为10-羰基十六烷酸,分析的水解产物分别为正己醇和9-羟基壬酸,其中,正己醇的分析条件为80℃保温2.5min,以10℃/min速率升温到110℃;9-羟基壬酸的分析条件为初始温度150℃,以5℃/min的升温速率升到215℃。
实施例8:底物为10-羰基十七烷酸,分析的水解产物分别为正庚醇和9-羟基壬酸,其中,正庚醇的分析条件为90℃保温8min;9-羟基壬酸的分析条件参照实施例8。
实施例9:底物为10-羰基十八烷酸,分析的水解产物分别为正辛醇和9-羟基壬酸,其中,正辛醇的分析条件为90℃保温8min;9-羟基壬酸的分析条件参照实施例8。
实施例10:底物为9-羰基十八烷酸,分析的水解产物分别为正壬醇和癸酸,其中,正壬醇的分析条件为初始温度80℃,以2℃/min的升温速率升到90℃,保温4min;癸酸的分析条件为初始温度120℃,以20℃/min的升温速率升到260℃,保温1min。
实施例11:底物为10-羰基十九烷酸,分析的水解产物分别为正壬醇和9-羟基壬酸,其中,正壬醇的分析条件为初始温度80℃,以2℃/min的升温速率升到90℃,保温4min;9-羟基壬酸的分析条件参照实施例8。
实施例12:底物为10-羰基二十烷酸,分析的水解产物分别为正癸醇和9-羟基壬酸,其中,正癸醇的分析条件为100℃保温10min;9-羟基壬酸的分析条件参照实施例8。
气相色谱分析结果见表2。
表2重组单加氧酶PaBVMON56L/S141L/F257Q/A310C/T313V催化长链脂肪酸Baeyer-Villiger单加氧反应结果
实施例13重组单加氧酶PaBVMON56L/S141L/F257Q/A310C/T313V催化制备癸二酸
反应在1L三口烧瓶中进行,在500mL甘氨酸-氢氧化钠缓冲液(100mmol/L,pH 9.0)中加入6250U如实施例6制备的PaBVMON56L/S141L/F257Q/A310C/T313V冻干酶粉和6250U葡萄糖脱氢酶冻干酶粉,加入150mg的10-羰基十八烷酸,再加入0.45g的葡萄糖和终浓度为0.1mmol/L的NADP+。20℃,200rpm机械搅拌反应24h。反应结束,用20%H2SO4将pH调到2,用等体积乙酸乙酯萃取3遍,合并萃取液,旋转蒸发浓缩至500ml,加入等体积饱和食盐水洗涤两遍,除去部分水溶性杂质。洗涤后的有机相减压旋转蒸发除去溶剂,加入25mL浓度为1M的KOH溶液,于60℃搅拌,水解反应2h,反应结束将温度提高到90℃,用20%H2SO4调节至pH7左右,恢复至室温,此时溶液中析出的沉淀中的杂质以脂肪酸盐等为主。悬浮液过滤,将滤液升温到90℃,用20%H2SO4调节至pH 2左右,恢复至室温,此时溶液中析出的沉淀是癸二酸粗品。过滤,向滤饼中加入4mL沸水使完全溶解,待自然冷却至室温后置于4℃过夜,过滤得到高纯度的癸二酸,置于105℃烘箱干燥,称重57.8mg,得率56.9%,核磁共振分析结构正确。
实施例14重组单加氧酶PaBVMON56L/S141L/F257Q/A310C/T313V催化制备壬二酸
反应在1L三口烧瓶中进行,在500mL甘氨酸-氢氧化钠缓冲液(100mmol/L,pH 9.0)中加入6250U如实施例6制备的PaBVMON56L/S141L/F257Q/A310C/T313V冻干酶粉和6250U葡萄糖脱氢酶冻干酶粉,加入150mg的9-羰基十八烷酸,再加入0.45g的葡萄糖和终浓度为0.1mmol/L的NADP+。20℃,200rpm机械搅拌反应24h。反应结束,用20%H2SO4将pH调到2,用等体积乙酸乙酯萃取3遍,合并萃取液,旋转蒸发浓缩至500ml,加入等体积饱和食盐水洗涤两遍,除去部分水溶性杂质。洗涤后的有机相减压旋转蒸发除去溶剂,加入25mL浓度为1M的KOH溶液,于60℃搅拌,水解反应2h,反应结束将温度提高到90℃,用20%H2SO4调节至pH7左右,恢复至室温,此时溶液中析出的沉淀中的杂质以脂肪酸盐等为主。悬浮液过滤,将滤液升温到90℃,用20%H2SO4调节至pH 2左右,恢复至室温,此时溶液中析出的沉淀是壬二酸粗品。过滤,向滤饼中加入4mL沸水使完全溶解,待自然冷却至室温后置于4℃过夜,过滤得到高纯度的壬二酸,置于105℃烘箱干燥,称重67.8mg,得率71.8%,核磁共振分析结构正确。
对比实施例1重组单加氧酶PaBVMO与单加氧酶WP_003087250.1的活性比较
将本发明所述单加氧酶PaBVMO与NCBI数据库中公开的预测为单加氧酶的蛋白质(登录号:WP_003087250.1)进行序列比对,其氨基酸序列一致性为99.0%。其中,本发明所述单加氧酶PaBVMO的第45位氨基酸残基为谷氨酸,而蛋白质WP_003087250.1的相应位点为天冬氨酸;PaBVMO的第106位氨基酸残基为丝氨酸,而蛋白质WP_003087250.1的相应位点为苏氨酸;PaBVMO的第226位氨基酸残基为天冬氨酸,而蛋白质WP_003087250.1的相应位点为谷氨酸;PaBVMO的第285位氨基酸残基为精氨酸,而蛋白质WP_003087250.1的相应位点为赖氨酸;PaBVMO的第362位氨基酸残基为半胱氨酸,而蛋白质WP_003087250.1的相应位点为甘氨酸。
重组单加氧酶PaBVMO对10-羰基十八烷酸的活性为34.6U/g,区域选择性为89:11。通过易错PCR随机突变获得突变体PaBVMOS141L,对该底物活性提高至121.4U/g,区域选择性提高至90:10,通过定点突变改造获得突变体PaBVMOS141L/F257Q/A310C和PaBVMON56L/S141L/F257Q/A310C/T313V,对该底物活性分别提高至247.9和366.8U/g,区域选择性分别提高至90:10和94:6。而已公开序列的蛋白质WP_003087250.1对10-羰基十八烷酸的活性和区域选择性分别为25.7U/g和78:22,结果表明已公开序列的蛋白质WP_003087250.1的活性测定和区域选择性均低于本发明所述的单加氧酶PaBVMO,更低于本发明所述的单加氧酶PaBVMO的突变体。
上述的对实施例的描述是为便于该技术领域的普通技术人员能理解和使用发明。熟悉本领域技术的人员显然可以容易地对这些实施例做出各种修改,并把在此说明的一般原理应用到其他实施例中而不必经过创造性的劳动。因此,本发明不限于上述实施例,本领域技术人员根据本发明的揭示,不脱离本发明范畴所做出的改进和修改都应该在本发明的保护范围之内。
序列表
<110> 华东理工大学;苏州百福安酶技术有限公司
<120> Baeyer-Villiger单加氧酶、突变体及其在制备中长链二元羧酸中的应用
<160> 12
<170> SIPOSequenceListing 1.0
<210> 1
<211> 1545
<212> DNA
<213> 铜绿假单胞菌(Pseudomonas aeruginosa)
<400> 1
atgagtaccc aacccacccc tgccgccgcc cggcactgca aggtcgccat catcggcacc 60
ggtttctccg ggctggggat ggcgatccgt ctccgccagg aaggcgagga cgacttcctg 120
attttcgaaa aggaagccgg cgtcggcggc acctggagag tcaacaacta ccctggctgc 180
gcctgcgacg tgcaatccca cgtctattcc ttctccttcg aagcgaaccc ggagtggacg 240
cggatgttcg cccgccagcc ggaaatccgc gcctatctgg agaagtgctg ggaaaaatat 300
cgcttgcagg aaaagtccct gctgaacacc gagatcggca aactggcctg ggacgagcgg 360
caaagcctct ggcacctgca tgatgcccag ggcaaccatt acaccgcgaa cgccgtggtt 420
tccggtatgg gcggcctgtc caccccggcc tatccgcgcc tcgatggcct ggagaacttc 480
cagggcaagg tcttccattc ccagcagtgg gaccatgact atgacctcaa gggcaagcgc 540
gtggcggtga tcggcaccgg cgcctcggcg atccagttcg tcccggagat ccagccgctg 600
gtggccgcgc tcgatctcta ccagcgcact ccgccatgga tcctgcccaa acccgaccgg 660
gcgattagcg aaaccgaccg ccggcgcttc cggcgcttcc cgctggtgca aaagctctgg 720
cgcggcggcc tctacagcct gctggaaggg cgggtgctgg gcttcacctt cgccccgcag 780
gtgatgaagc tggtgcagcg cctggcgatc cgccacatcc acaagcagat caaggatccg 840
gaactgcgcc gccgcgtcac gccggactac accatcggct gcaagcgcat cctcatgtcg 900
cacaactact atccggccct ggctgccgcc aattccacgg tgatcaccga aggcatccgc 960
gccgtcaccg ccaacggaat cgtcgacggc aacggccggg aacgcgaggt cgacgcgatt 1020
attttcggta ccggcttcac cgccaacgac cctatccccc gcggagtggt attcggtcgc 1080
gactgtcgcg acctgctgga cagctggacc aagggcccgg aagcctacaa gggcactacc 1140
accgccggct tccccaacct gttcttcctg atgggaccga ataccggcct cggccacaac 1200
tccatggtct acatgatcga gtcgcagatc gcctacgtcc tcgatgcgct gaagctgatg 1260
aagcgccgcg aactgctcag tctcgaggtc aaagccccgg tgcaggaacg ctacaacgaa 1320
tacctccagc gcaagctgga ccgcagcgtc tggagcgtgg gcggttgcaa gagctggtat 1380
ctgcatccgg tcagcggccg caactgcacc ctgtggccgg gattcacctg gcgcttccgc 1440
gctctgaccc ggcagttcga cgcctccgcc taccacctca ccacgacacc gctcgccgct 1500
ctaagcaacg aagcccgcca acaggccgaa ggggtacccg catga 1545
<210> 2
<211> 514
<212> PRT
<213> 铜绿假单胞菌(Pseudomonas aeruginosa)
<400> 2
Met Ser Thr Gln Pro Thr Pro Ala Ala Ala Arg His Cys Lys Val Ala
1 5 10 15
Ile Ile Gly Thr Gly Phe Ser Gly Leu Gly Met Ala Ile Arg Leu Arg
20 25 30
Gln Glu Gly Glu Asp Asp Phe Leu Ile Phe Glu Lys Glu Ala Gly Val
35 40 45
Gly Gly Thr Trp Arg Val Asn Asn Tyr Pro Gly Cys Ala Cys Asp Val
50 55 60
Gln Ser His Val Tyr Ser Phe Ser Phe Glu Ala Asn Pro Glu Trp Thr
65 70 75 80
Arg Met Phe Ala Arg Gln Pro Glu Ile Arg Ala Tyr Leu Glu Lys Cys
85 90 95
Trp Glu Lys Tyr Arg Leu Gln Glu Lys Ser Leu Leu Asn Thr Glu Ile
100 105 110
Gly Lys Leu Ala Trp Asp Glu Arg Gln Ser Leu Trp His Leu His Asp
115 120 125
Ala Gln Gly Asn His Tyr Thr Ala Asn Ala Val Val Ser Gly Met Gly
130 135 140
Gly Leu Ser Thr Pro Ala Tyr Pro Arg Leu Asp Gly Leu Glu Asn Phe
145 150 155 160
Gln Gly Lys Val Phe His Ser Gln Gln Trp Asp His Asp Tyr Asp Leu
165 170 175
Lys Gly Lys Arg Val Ala Val Ile Gly Thr Gly Ala Ser Ala Ile Gln
180 185 190
Phe Val Pro Glu Ile Gln Pro Leu Val Ala Ala Leu Asp Leu Tyr Gln
195 200 205
Arg Thr Pro Pro Trp Ile Leu Pro Lys Pro Asp Arg Ala Ile Ser Glu
210 215 220
Thr Asp Arg Arg Arg Phe Arg Arg Phe Pro Leu Val Gln Lys Leu Trp
225 230 235 240
Arg Gly Gly Leu Tyr Ser Leu Leu Glu Gly Arg Val Leu Gly Phe Thr
245 250 255
Phe Ala Pro Gln Val Met Lys Leu Val Gln Arg Leu Ala Ile Arg His
260 265 270
Ile His Lys Gln Ile Lys Asp Pro Glu Leu Arg Arg Arg Val Thr Pro
275 280 285
Asp Tyr Thr Ile Gly Cys Lys Arg Ile Leu Met Ser His Asn Tyr Tyr
290 295 300
Pro Ala Leu Ala Ala Ala Asn Ser Thr Val Ile Thr Glu Gly Ile Arg
305 310 315 320
Ala Val Thr Ala Asn Gly Ile Val Asp Gly Asn Gly Arg Glu Arg Glu
325 330 335
Val Asp Ala Ile Ile Phe Gly Thr Gly Phe Thr Ala Asn Asp Pro Ile
340 345 350
Pro Arg Gly Val Val Phe Gly Arg Asp Cys Arg Asp Leu Leu Asp Ser
355 360 365
Trp Thr Lys Gly Pro Glu Ala Tyr Lys Gly Thr Thr Thr Ala Gly Phe
370 375 380
Pro Asn Leu Phe Phe Leu Met Gly Pro Asn Thr Gly Leu Gly His Asn
385 390 395 400
Ser Met Val Tyr Met Ile Glu Ser Gln Ile Ala Tyr Val Leu Asp Ala
405 410 415
Leu Lys Leu Met Lys Arg Arg Glu Leu Leu Ser Leu Glu Val Lys Ala
420 425 430
Pro Val Gln Glu Arg Tyr Asn Glu Tyr Leu Gln Arg Lys Leu Asp Arg
435 440 445
Ser Val Trp Ser Val Gly Gly Cys Lys Ser Trp Tyr Leu His Pro Val
450 455 460
Ser Gly Arg Asn Cys Thr Leu Trp Pro Gly Phe Thr Trp Arg Phe Arg
465 470 475 480
Ala Leu Thr Arg Gln Phe Asp Ala Ser Ala Tyr His Leu Thr Thr Thr
485 490 495
Pro Leu Ala Ala Leu Ser Asn Glu Ala Arg Gln Gln Ala Glu Gly Val
500 505 510
Pro Ala
<210> 3
<211> 27
<212> DNA
<213> 人工序列(Artificial Sequence)
<400> 3
ccggaattca tgagtaccca acccacc 27
<210> 4
<211> 27
<212> DNA
<213> 人工序列(Artificial Sequence)
<400> 4
cccaagcttt catgcgggta ccccttc 27
<210> 5
<211> 33
<212> DNA
<213> 人工序列(Artificial Sequence)
<400> 5
ggcttcaccc aagccccgca ggtgatgaag ctg 33
<210> 6
<211> 33
<212> DNA
<213> 人工序列(Artificial Sequence)
<400> 6
ctgcggggct tgggtgaagc ccagcacccg ccc 33
<210> 7
<211> 33
<212> DNA
<213> 人工序列(Artificial Sequence)
<400> 7
ctggctgcct gcaattccac ggtgatcacc gaa 33
<210> 8
<211> 33
<212> DNA
<213> 人工序列(Artificial Sequence)
<400> 8
cgtggaattg caggcagcca gggccggata gta 33
<210> 9
<211> 33
<212> DNA
<213> 人工序列(Artificial Sequence)
<400> 9
agagtcaacc tttaccctgg ctgcgcctgc gac 33
<210> 10
<211> 33
<212> DNA
<213> 人工序列(Artificial Sequence)
<400> 10
gccagggtaa aggttgactc tccaggtgcc gcc 33
<210> 11
<211> 33
<212> DNA
<213> 人工序列(Artificial Sequence)
<400> 11
gccaattccg ttgtgatcac cgaaggcatc cgc 33
<210> 12
<211> 33
<212> DNA
<213> 人工序列(Artificial Sequence)
<400> 12
ggtgatcaca acggaattgg cggcagccag ggc 33
- 还没有人留言评论。精彩留言会获得点赞!