一种高产L-苹果酸的黑曲霉基因工程菌株及其构建与应用的制作方法
本发明属于生物工程领域,更具体地说涉及一种高产l-苹果酸的黑曲霉基因工程菌株的构建及应用。
背景技术:
l-苹果酸,又名2-羟基丁二酸,主要用于食品、医药等行业。在食品行业,由于l-苹果酸味道柔和、酸度大,且不损害口腔牙齿、不积累脂肪,成为一种低热量的国际食品界公认的安全性食品酸味剂,是目前世界食品行业中用量最大和发展前景较好的有机酸之一。在医药行业,l-苹果酸被用于治疗肝病、贫血、尿毒症等多种疾病。而且由于l-苹果酸在代谢上利于氨基酸的吸收,常被配入复合氨基酸注射液中。此外,l-苹果酸在日化保健业等行业也有广泛的应用前景。因此,国际市场上对l-苹果酸的需求量与日俱增。
当前,化学合成法和固定化酶技术是工业上生产苹果酸的主要方法。其中,化学合成法生产的苹果酸为消旋型dl-苹果酸,而一些国家规定饮料和药品中不能使用dl-苹果酸,限制了消旋型苹果酸的应用范围。固定化酶技术虽然可以光学纯度的l-苹果酸,但是其底物富马酸来源于石油基化学品马来酸。因此,具有较好经济、环境和社会效益的发酵法生产l-苹果酸成为研究的热点。然而,目前发酵法生产苹果酸存在的问题仍是缺乏优良的生产菌株。利用黄曲霉菌株发酵合成l-苹果酸,虽然能够获得113g/l的产量,但是由于产生黄曲霉毒素和高浓度杂酸,限制了其工业应用。近年来应用基因工程策略,对大肠杆菌、酵母菌等微生物代谢途径进行改造获得基因工程菌株取得较大进展,但是由于产量不够高或者有大量副产物杂酸的存在等瓶颈,阻碍了其工业化发酵生产苹果酸的进程。
黑曲霉的优势:黑曲霉作为重要的细胞工厂用于柠檬酸发酵生产已有100多年的历史,具有非常强的耐酸性,在ph低于2的条件下能够正常生长,不仅是gras(generallyregardedsafe)菌株,而且能够利用廉价的碳源。然而由于野生型黑曲霉菌株较低的苹果酸产量,鲜有研究报道通过代谢工程策略,改造黑曲霉菌种用于苹果酸的发酵生产。
技术实现要素:
本发明的目的在于提供一种高产l-苹果酸的黑曲霉基因工程菌株。
一种高产l-苹果酸的黑曲霉基因工程菌株,所述黑曲霉基因工程菌株是过表达黑曲霉来源的丙酮酸羧化酶基因pyc和黑曲霉来源的苹果酸脱氢酶基因mdh、异源表达米曲霉来源的四碳二羧酸转运蛋白c4t318基因并敲除了草酰乙酸水解酶基因oaha的黑曲霉基因工程菌株。
本发明所用的宿主菌株是本实验室构建的能够表达cre重组酶的黑曲霉菌株s469。所述s469菌株是在基因组内整合了外源cre基因,该基因受tet-on系统的调控表达。当以所述菌株s469为出发菌进行遗传改造,并以loxp-hph-loxp为筛选标记时,可通过强力霉素启动tet-on系统表达cre重组酶,实现对loxp-hph-loxp元件的重组,从而实现应用一个hphmarker进行连续的基因过表达或敲除且实现最终目的工程菌株基因组内无外源抗性基因的残留。
所述黑曲霉基因工程菌株是过表达黑曲霉来源的丙酮酸羧化酶基因pyc和黑曲霉来源的苹果酸脱氢酶基因mdh、异源表达米曲霉来源的四碳二羧酸转运蛋白c4t318基因并敲除了草酰乙酸水解酶基因oaha的黑曲霉基因工程菌株。
本发明所述基因pyc序列在ncbi-locus_tag是an04g02090,所述基因mdh序列在ncbi-geneid是4987622,所述c4t318基因在ncbi-geneid是5992883,所述基因oaha序列在ncbi-locus_tag是an10g00820。
为实现本发明的目的,依次通过以下步骤实现:
(1)cre-loxp遗传操作系统的构建
首先构建尿嘧啶营养缺陷菌株s296,然后将tet-on调控cre表达的质粒plh409(tet-on-cre,pyrg)转化至s296,筛选得到强力霉素诱导表达cre的黑曲霉菌株s469。因此可以实现对loxp-hph-loxp元件中潮霉素抗性基因hph的重组去除,从而可以反复使用hphmarker实现连续的对多个基因的过表达或敲除。
(2)高产l-苹果酸的黑曲霉基因工程菌株的构建
步骤1,构建pyc基因表达质粒:以野生型黑曲霉atcc1015基因组为模板,通过pcr反应扩增获得基因pyc序列片段。将所述基因pyc序列片段克隆到载体plh331,构建基因pyc表达质粒plh395。
步骤2,pyc基因过表达菌株的获得:将所述质粒plh395转化至宿主菌株s469,经转化子筛选和潮霉素抗性基因重组获得pyc基因过表达菌株。
步骤3,构建表达mdh基因质粒:以野生型黑曲霉atcc1015基因组为模板,通过pcr反应扩增获得基因mdh序列片段。将所述基因mdh序列片段克隆到载体plh331,构建基因mdh表达质粒plh373。
步骤4,pyc基因和mdh基因共过表达菌株的获得:将所述质粒plh373转化至pyc基因过表达菌株s453,经转化子筛选和潮霉素抗性基因重组获得pyc基因和mdh基因共过表达菌株。
步骤5,构建表达c4t318基因质粒:以野生型米曲霉nrrl3488基因组为模板,通过pcr反应扩增获得基因c4t318序列片段。将所述基因c4t318序列片段克隆到载体plh454,构建基因c4t318表达质粒plh455。所述基因c4t318由黑曲霉3-磷酸甘油脱氢酶基因启动子pgpda控制,所述启动子pgpda序列为序列表中的seqno.4,长度为932bp。
步骤6,pyc和mdh基因共过表达且异源表达c4t318基因菌株的获得:将所述质粒plh455转化至pyc和mdh基因共过表达菌株,经转化子筛选和潮霉素抗性基因重组获得pyc和mdh基因共过表达且异源表达c4t318基因菌株s1。
步骤7,构建基因oaha敲除质粒:以野生型黑曲霉atcc1015基因组为模板,通过pcr反应分别扩增获得基因oaha的上游和下游序列片段。将所述基因oaha的上下游序列片段克隆到载体plh314,构建基因oaha敲除质粒plh398。
步骤8,pyc和mdh基因共过表达、异源表达c4t318基因且oaha基因敲除菌株的获得:将所述质粒plh398转化至pyc和mdh基因共过表达且异源表达c4t318基因菌株,经转化子筛选和潮霉素抗性基因重组获得pyc基因和mdh基因共过表达、异源表达c4t318基因且oaha基因敲除菌株s2。
本发明还提供上述基因重组菌株在制备苹果酸中的应用。
利用本发明构建的黑曲霉重组菌株发酵产生苹果酸的方法,具体步骤如下:
首先,将菌株接种在pda培养平板上在28℃培养6天直至产生分生孢子。
然后,将孢子粉接种中摇瓶发酵培养基中,孢子的终浓度为2×106孢子/ml,在28℃,220rpm培养7天。
发酵培养基的组成为:100g/lglucose,80g/lcaco3,6g/lbactopeptone,150mg/lkh2po4,150mg/lk2hpo4,100mg/lmgso·7h2o,100mg/lcacl2·2h2o,5mg/lfeso4·7h2o,5mg/lnacl。
本发明的有益效果是:
本发明基于黑曲霉产生苹果酸的天然特性,通过遗传重组改造黑曲霉的生理代谢途径,利用基因表达及敲除技术对其进行改造,获得了一种黑曲霉基因工程菌株,经过实验证实,该黑曲霉基因工程菌株产生苹果酸的能力显著提升,摇瓶发酵结束后苹果酸产量达到120±2.8g/l且没有副产物草酸产生。为微生物发酵法制备苹果酸提供了优良菌种。
附图说明
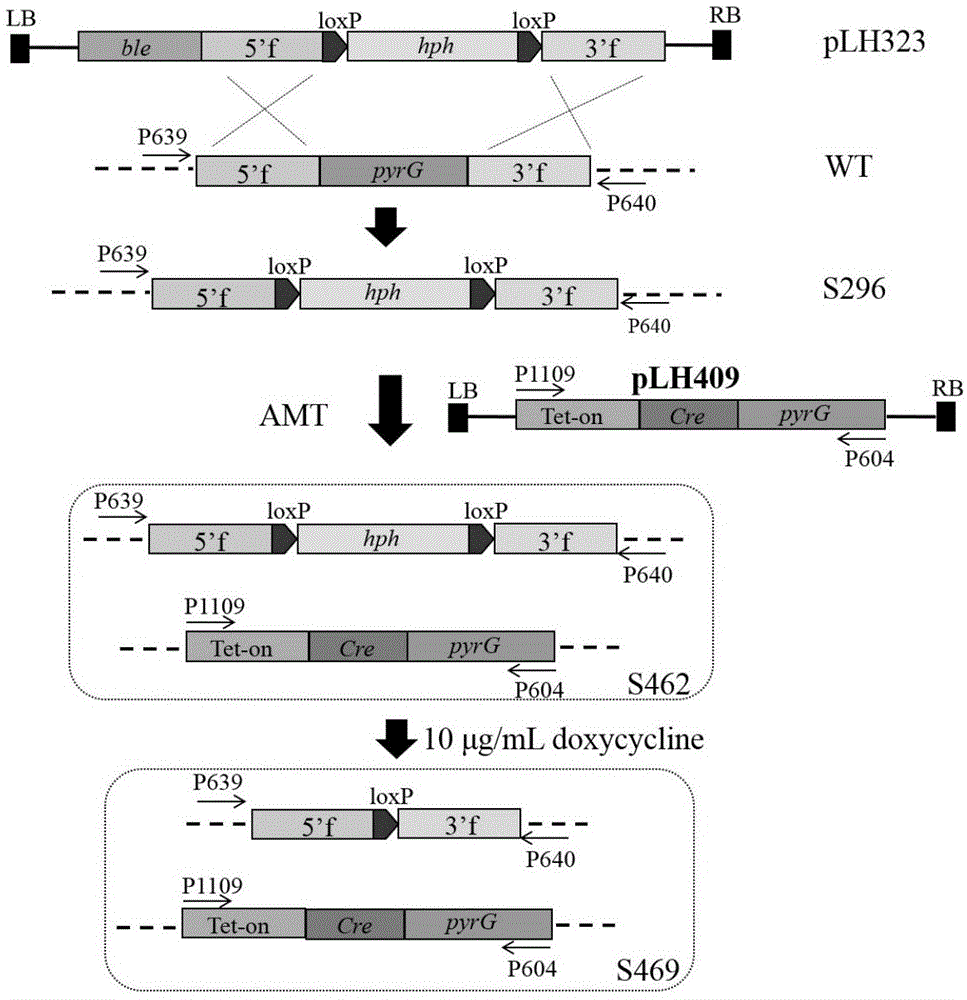
图1是本发明中cre表达菌株s469的构建流程。amt表示农杆菌介导转化。
图2是本发明中构建的pyc表达质粒plh395图谱。
图3是本发明中对pyc表达质粒plh395双酶切验证图,其中m为dnamarker,1和2为spei和hindiii双酶切验证质粒。
图4是本发明中构建的mdh表达质粒plh373图谱。
图5是本发明中对pyc表达质粒plh373双酶切验证图,其中m为dnamarker,1为ecori和bamhi双酶切验证质粒。
图6是本发明中构建的c4t318表达质粒plh455图谱。
图7是本发明中对c4t318表达质粒plh455双酶切验证图,其中m为dnamarker,1和2为spei和xbaiii双酶切验证质粒。
图8是本发明中构建的oaha敲除质粒plh398图谱。
图9是本发明中构建的oaha敲除质粒plh398双酶切验证图,其中m为dnamarker,1为ecori和bamhi双酶切验证上游同源臂质粒,2为spei和hindiii双酶切验证下游同源臂质粒。
图10是本发明中基因工程菌株发酵液的高效液相色谱分析图。s1为pyc和mdh基因共过表达且异源表达c4t318基因菌株的发酵液高效液相色谱分析图,ss2为pyc和mdh基因共过表达、异源表达c4t318基因且oaha基因敲除菌株s2的发酵液高效液相色谱分析图。红色方框内为草酸峰图。
图11是本发明中各工程菌株的苹果酸产量。s469为出发菌株在第7天的苹果酸产量,s1为pyc和mdh基因共过表达且异源表达c4t318基因菌株在第7天的苹果酸产量,s2为pyc和mdh基因共过表达、异源表达c4t318基因且oaha基因敲除菌株在第7天的苹果酸产量。
具体实施方式
下面通过具体实施例对本发明的技术方案作进一步详细说明。
实施例1
本实施例包括以下步骤:
(1)含有loxp-hph-loxp元件质粒的构建
loxp-hph-loxp元件序列由北京华大基因合成,然后经bamhi和psti酶切连接克隆到经过相同双酶切的载体pfgl59ble构建质粒plh314(loxp-hph-loxp,bler)。plh314经xhoi单酶切后连接得到质粒plh331(loxp-hph-loxp)。plh314用于构建敲除质粒的出发载体;以黑曲霉atcc1015基因组为模板,p635/p636和p637/p638为引物分别扩增pyrg基因两端的dna序列,然后分别克隆到载体plh314构建pyrg敲除质粒plh323。
(2)含有tet-on-cre元件质粒的构建
sali单酶切载体pfgl59将hph抗性基因去除,回收载体片段后经dnat4连接酶连接获得plh309。以黑曲霉atcc1015基因组为模板,p603/p604为引物经pcr扩增pyrg及其自身启动子和终止子,经smai和xbai双酶切后连接到经相同双酶切的质粒plh309得到质粒plh310(pyrg)。cre经密码子优化后,tet-on-cre序列由北京华大基因合成,然后经spei和ncoi双酶切后连接到经相同双酶切的载体plh310(pyrg)得到tet-on调控cre表达的质粒plh409(tet-on-cre,pyrg)。pyrg扩增产物经华大基因公司测序验证未有突变。
(3)cre-loxp遗传操作系统的构建
为了将cre表达元件成功整合到黑曲霉基因组且不残留外源的抗性标记基因,因此选用pyrg为筛选标记进行转化。以pyrg为筛选标记进行转化的前提是出发菌株为pyrg缺陷菌株,即尿嘧啶营养缺陷菌株δpyrg。因此首先通过农杆菌介导转化的方法将plh323转化到黑曲霉野生菌株atcc1015中,经双交换同源重组得到pyrg敲除菌株δpyrgs296。在同源臂以外的序列上设计引物p639/p640,提取s296基因组为模板,经pcr验证pyrg基因被敲除。
由于pyrg部分序列被loxp-hph-loxp取代,得到的转化子具有尿嘧啶营养缺陷和潮霉素抗性的特点,因此s296必须在补充有尿嘧啶的培养基中培养。
以δpyrgs296为出发菌株,通过农杆菌介导转化质粒plh409,将由tet-on调控表达的cre基因整合到s296基因组得到菌株s462。由于plh409转化过程是以pyrg为筛选标记,所以得到的阳性转化子中pyrg基因得以回补成为尿嘧啶原养型,即出发菌株的尿嘧啶营养缺陷表型得以恢复。提取s462基因组,以p1109/p604为引物,经pcr验证tet-on-cre元件是否整合到基因组。
s462菌株中tet-on-cre元件的成功整合,使得s462菌株在含有强力霉素(doxycycline)的培养基中能够诱导表达cre重组酶,cre重组酶可以识别loxp位点,从而将loxp-hph-loxp中的hphmarker重组掉,实现hphmarker的反复使用,进而连续应用同一个hphmarker实现多个基因的过表达或敲除。
为了重组去除s462菌株中的hphmarker,将黑曲霉s462的孢子约300个,均匀涂布在含有10μg/ml强力霉素的mm培养基(碳源为葡萄糖)中进行诱导培养至长出单克隆,挑取100个单克隆同时接种于pda和含有250μg/ml潮霉素的pda培养基中,在含有250μg/ml潮霉素的pda培养基中筛选潮霉素敏感克隆,用p607/p608引物pcr验证hphmarker的去除,选择其中一个正确的克隆,命名为s469。该s469菌株即为构建高产l-苹果酸的黑曲霉基因工程菌株的宿主菌。
实施例2
本实施例包括以下步骤:
(1)pyc表达质粒的构建
为扩增pyc基因序列,设计引物p994/p995(表1),通过pcr扩增得到pyc基因序列片段,送至华大基因公司测序确认无突变,然后经spei和hindiii双酶切回收后与同样内酶切处理的质粒片段plh331进行连接,将连接产物转化于大肠杆菌jm109感受态细胞,并均匀涂布于含有100μg/ml卡那霉素的lb培养皿中,37℃过夜培养,挑取单克隆,经菌落pcr验证和双酶切验证(图2),获得pyc表达质粒plh395。
所述基因pyc序列包含该基因自身的启动子和终止子,为序列表中的seqno.1,长度为5247bp。
(2)mdh表达质粒的构建
为扩增mdh基因序列,设计引物p819/p820(表1),通过pcr扩增得到mdh基因序列片段,送至华大基因公司测序确认无突变,经ecori和bamhi双酶切回收后与同样内酶切处理的质粒片段plh331进行连接,将连接产物转化于大肠杆菌jm109感受态细胞,并均匀涂布于含有100μg/ml卡那霉素的lb培养皿中,37℃过夜培养,挑取单克隆,经菌落pcr验证和双酶切验证(图4),获得mdh表达质粒plh373。
所述基因mdh序列包含该基因自身的启动子和终止子,为序列表中的seqno.2,长度为2849bp。
(3)c4t318表达质粒的构建
为扩增c4t318基因序列,设计引物p1176/p1177(表1),通过pcr扩增得到c4t318基因序列片段,送至华大基因公司测序确认无突变,经ecori和kpni双酶切回收后与同样内酶切处理的质粒片段plh454进行连接,将连接产物转化于大肠杆菌jm109感受态细胞,并均匀涂布于含有100μg/ml卡那霉素的lb培养皿中,37℃过夜培养,挑取单克隆,经菌落pcr验证和双酶切验证(图6),获得c4t318表达质粒plh373。
所述基因c4t318序列起始于起始密码子atg,包含该基因编码序列和自身终止子,为序列表中的seqno.3,长度为1411bp。
(4)oaha敲除质粒的构建
为扩增oaha上游序列片段,设计引物p953/p954(表1),通过pcr扩增得到oaha上游序列片段,经ecori和bamhi双酶切回收后与相同内酶切处理的质粒片段plh314进行连接,将连接产物转化于大肠杆菌jm109感受态细胞,并均匀涂布于含有100μg/ml卡那霉素的lb培养皿中,37℃过夜培养,挑取单克隆,经菌落pcr验证和双酶切验证(图8),获得质粒plh314::oaha-5’f。所述基因oaha的上游序列为序列表中的seqno.5,长度为1094bp。
为扩增oaha下游序列片段,设计引物p955/p956(表1),通过pcr扩增得到oaha下游序列片段,经spei和hindiii双酶切回收后与同样内酶切处理的质粒片段plh314::oaha-5’f进行连接,将连接产物转化于大肠杆菌jm109感受态细胞,并均匀涂布于含有100μg/ml卡那霉素的lb培养皿中,37℃过夜培养,挑取单克隆,经菌落pcr验证和双酶切验证(图4),获得oaha敲除质粒plh398。所述基因oaha的下游序列为序列表中的seqno.6,长度为1235bp。
(5)以上所述lb培养基组分:
胰蛋白胨10.0g/l,酵母浸出物5.0g/l,nacl10.0g/l,ph调至7.0-7.2,固体培养基加1.5%(w/t)的琼脂粉。121℃灭菌20min。灭菌完毕冷却至60℃左右时加入卡那霉素至终浓度100μg/ml。
表1所用引物序列
a下划线序列表示酶切位点。
实施例3
本实施例包括以下步骤:
农杆菌介导黑曲霉转化及克隆筛选:本发明所述过表达或异源表达,是将相关基因整合到黑曲霉基因组进行表达。本发明所述表达基因及敲除基因的转化方法为农杆菌介导法(chenetal.2014,参考文献1)。所述农杆菌为agl-1菌株。本发明所述表达基因及敲除基因在农杆菌介导转化黑曲霉之前,需将所述表达质粒和敲除质粒首先电转化至农杆菌。所述电转条件是:capacitnce:25uf,voltage:2.5kv,resistance:200ω,pulse:5msec。
(1)pyc基因过表达菌株的获得
将质粒plh395电转至农杆菌,然后将含有质粒plh395的农杆菌与黑曲霉宿主菌株s469在mm平板共培养进行农杆菌介导转化,共培养两天后将转化子转接于含有200μm头孢噻肟,100μg/ml氨苄青霉素,100μg/ml链霉素,250μg/ml潮霉素b的cm平板中进行筛选直至转化子长出菌丝,然后随机挑取20个转化子进行摇瓶发酵筛选,选取产量最高的转化子进行hphmarker诱导重组。所述诱导重组方法为:将约300个转化子孢子均匀涂布与含有10μg/ml强力霉素的mm平板中至长出克隆,然后随机挑取100个克隆同时转接至pda平板和含有潮霉素的pda平板中,在含潮霉素的pda平板中不能生长而在pda能正常生长的克隆即为hphmarker诱导重组,表现为潮霉素敏感,该菌株即为pyc基因过表达菌株。
(2)pyc基因和mdh基因共过表达菌株的获得
将质粒plh373电转至农杆菌,然后将含有plh373的农杆菌与pyc基因过表达菌株在mm平板共培养进行农杆菌介导转化,共培养两天后将转化子转接于含有200μm头孢噻肟,100μg/ml氨苄青霉素,100μg/ml链霉素,250μg/ml潮霉素b的cm平板中进行筛选直至转化子长出菌丝,然后随机挑取20个转化子进行摇瓶发酵筛选,选取产量最高的转化子进行hphmarker诱导重组,获得潮霉素敏感的pyc基因和mdh基因共过表达菌株。所述诱导重组方法同上。
(3)pyc和mdh基因共过表达且异源表达c4t318基因菌株的获得
将质粒plh455电转至农杆菌,然后将含有质粒plh455的农杆菌与pyc基因和mdh基因共过表达菌株在mm平板共培养进行农杆菌介导转化,共培养两天后将转化子转接于含有200μm头孢噻肟,100μg/ml氨苄青霉素,100μg/ml链霉素,250μg/ml潮霉素b的cm平板中进行筛选直至转化子长出菌丝,然后随机挑取20个转化子进行摇瓶发酵筛选,选取产量最高的转化子进行hphmarker诱导重组,获得潮霉素敏感的pyc和mdh基因共过表达且异源表达c4t318基因菌株s1。所述诱导重组方法同上。
(4)pyc和mdh基因共过表达、异源表达c4t318基因且oaha基因敲除菌株的获得
将质粒plh398电转至农杆菌,然后将含有质粒plh398的农杆菌与pyc和mdh基因共过表达且异源表达c4t318基因菌株在mm平板共培养进行农杆菌介导转化,共培养两天后将转化子转接于含有200μm头孢噻肟,100μg/ml氨苄青霉素,100μg/ml链霉素,250μg/ml潮霉素b的cm平板中进行筛选直至转化子长出菌丝,然后随机挑取100个转化子转接至含有10μg/ml博来霉素的pda平板中,然后挑取在含有博来霉素的平板上生长缓慢的克隆提取基因组进行pcr筛选验证,验证引物为p973/p976(见表1)。由于敲除是双交换同源重组原理,oaha被loxp-hph-loxp替代。挑取其中一个正确的oaha敲除克隆进行hphmarker诱导重组从而获得不具有潮霉素抗性的pyc和mdh基因共过表达、异源表达c4t318基因且oaha基因敲除菌株s2。所述诱导重组方法同上。
(5)以上所述pda培养基组分:马铃薯200g,切成小块,加1000ml水煮沸30min,用双层纱布滤成清液。然后加入20g葡萄糖完全溶解,加水定容至1l。固体培养基加琼脂20g。121℃,20min高压灭菌。
(6)以上所述cm培养基组分:
20g琼脂,加水到897ml,121℃灭菌20min。微波加热待琼脂完全溶解后,加入:asp+n20ml,50%葡萄糖20ml,1mmgso42ml,cmtraceelements1ml,10%酪蛋白水解物10ml,10%酵母浸出物50ml。
所述cm培养基中所需试剂的配制:
1)asp+n:kcl(350mm)2.61g,kh2po4(550mm)7.48g,nano3(3.5m)29.75g,加入去离子水定容至100mlph5.5(5mkoh),121℃灭菌20min。
2)50%葡萄糖:葡萄糖50g,加入ddh2o定容至100ml,115℃灭菌20min。
3)1mmgso4:mgso424.648g,加入ddh2o定容至100ml,121℃灭菌20min。
4)cmtraceelements:znso4·7h2o(76mm)2.1g,h3bo3(178mm)1.1g,mncl2·4h2o(25mm)0.5g,feso4·7h2o(18mm)0.5g,cocl2·6h2o(7.1mm)0.17g,cuso4·5h2o(6.4mm)0.16g,na2moo4·2h2o(6.2mm)0.15g,edta(174mm)5.1g,加入去离子水定容至100ml,121℃灭菌20min。
5)10%酪蛋白水解物:酪蛋白水解物10g,加入ddh2o定容至100ml,121℃灭菌20min。
6)10%酵母浸出物:酵母浸出物10g,加入ddh2o定容至100ml,121℃灭菌20min。
(7)以上所述mm培养基组分:vogel'ssalts20ml,葡萄糖15g,琼脂15g,蒸馏水溶解并定容至1000ml。121℃灭菌20min。
所述mm培养基中所需试剂的配制:
1)vogel's50xsalts:柠檬酸钠150g,kh2po4250g,nh4no3100gmgso4·7h2010g,cacl2·2h205g。微量元素5ml,生物素溶液2.5ml,蒸馏水溶解并定容至1000ml,加0.2ml氯仿作为防腐剂,室温下保存。
2)微量元素溶液:柠檬酸·h205.00g,znso4·7h205.00g,fe(nh4)2(so4)2·6h201.00g,cuso4·5h200.25g,mnso4·h200.05g,h3bo30.05g,na2moo4·2h200.05g,蒸馏水溶解并定容至100ml,加1ml氯仿作为防腐剂,室温下保存。
3)生物素5.0mg,蒸馏水溶解并定容至50ml,-20℃保存。
实施例4:高产黑曲霉基因工程菌株发酵生产l-苹果酸
样品制备:摇匀发酵悬液,取1ml发酵液加入等体积的2mhcl溶解有机酸钙沉淀及残留的caco3,离心后再稀释50倍,经0.22μm滤膜过滤后滤液用于hplc检测。
苹果酸的测定方法:aminexhpx-87h柱(300mm×7.8mm),uv检测器。流动相:5mmh2so4。流速0.6ml/min,柱温65℃,波长210nm,进样体积为20μl。
分别将实施例1中获得宿主菌株s469和实施例3获得的黑曲霉基因工程菌株的分生孢子接种于发酵培养基中,28℃培养7天。经样品制备后,利用hplc法测定发酵液中l-苹果酸的含量。结果显示,经过7天摇瓶发酵,出发菌s469产量为16.2±3.0g/l,s1(pyc和mdh基因共过表达、异源表达c4t318基因)和s2(pyc和mdh基因共过表达、异源表达c4t318基因且oaha基因敲除菌株)产量分别为100.3±3.1g/l和120.4±2.8g/l(图10)。pyc和mdh基因共过表达、异源表达c4t318基因且oaha基因敲除菌株s2相比与出发菌s469提高了7.43倍。本发明中苹果酸相对于葡萄糖的转化率为1.62mol/mol,达到理论最高转化率的81%。
该菌发酵生产苹果酸的最适ph范围在6.0至7.0之间,因此发酵液中添加碳酸钙维持ph条件的稳定。发酵产生的苹果酸及副产物如柠檬酸、富马酸等有机酸主要以钙盐的形式存在于发酵液中。发酵过程的第一天主要是菌体的生长阶段,只有微量的苹果酸产生,发酵整个过程温度控制在28至30℃之间,菌体主要以较大菌球的形态存在。
本发明的研究成果证实了黑曲霉发酵生产苹果酸的潜力,为苹果酸的工业化生产提供了优良的菌株。
序列表
<110>天津科技大学
<120>一种高产l-苹果酸的黑曲霉基因工程菌株及其构建与应用
<160>22
<170>siposequencelisting1.0
<210>1
<211>53
<212>dna
<213>pyrg基因扩增引物p603(unknown)
<400>1
tcccccgggactagtgaattcctcgagccatggctcactgttcctttacggat53
<210>2
<211>28
<212>dna
<213>pyrg基因扩增引物p604(unknown)
<400>2
gctctagagctgacgctgacttggatgc28
<210>3
<211>29
<212>dna
<213>pyrg5’f同源臂扩增引物p635(unknown)
<400>3
ccggaattccttgcagacaatgccattct29
<210>4
<211>28
<212>dna
<213>pyrg5’f同源臂扩增引物p636(unknown)
<400>4
cgggatccattgggatgcttgctggcac28
<210>5
<211>29
<212>dna
<213>pyrg3’f同源臂扩增引物p637(unknown)
<400>5
gctctagagaggatcgaagttctgatggt29
<210>6
<211>29
<212>dna
<213>pyrg3’f同源臂扩增引物p638(unknown)
<400>6
cagggcccttctcatccgccatgttagat29
<210>7
<211>20
<212>dna
<213>pyrg基因敲除验证引物p639(unknown)
<400>7
agtcatagcagattcaagct20
<210>8
<211>20
<212>dna
<213>pyrg基因敲除验证引物p640(unknown)
<400>8
gtctgcatccttcgtatgct20
<210>9
<211>27
<212>dna
<213>mdh基因扩增引物p819(unknown)
<400>9
cgagctctccagaagtgactaagcaac27
<210>10
<211>28
<212>dna
<213>mdh基因扩增引物p820(unknown)
<400>10
cgggatcctacagtatacgttcatcact28
<210>11
<211>28
<212>dna
<213>oaha5’f同源臂扩增引物p953(unknown)
<400>11
cggaattcgagccctggcagtctatcgg28
<210>12
<211>33
<212>dna
<213>oaha5’f同源臂扩增引物p954(unknown)
<400>12
cgggatccagaaagaggcttgtttgagactgat33
<210>13
<211>32
<212>dna
<213>oaha3’f同源臂扩增引物p955(unknown)
<400>13
ggactagttttgtttcacccagcagaacctta32
<210>14
<211>29
<212>dna
<213>oaha3’f同源臂扩增引物p956(unknown)
<400>14
cccaagcttatcggcaaggagcgtcgtct29
<210>15
<211>26
<212>dna
<213>oaha基因敲除验证引物p973(unknown)
<400>15
gaaaactgggtgttagatttcagttg26
<210>16
<211>20
<212>dna
<213>oaha基因敲除验证引物p976(unknown)
<400>16
tgttctgccagccgttagga20
<210>17
<211>26
<212>dna
<213>pyc基因扩增引物p994(unknown)
<400>17
ggactagtagagtccgatgttgctgg26
<210>18
<211>28
<212>dna
<213>pyc基因扩增引物p995(unknown)
<400>18
ccaagcttcttctgaataaatggaggtt28
<210>19
<211>28
<212>dna
<213>tet-on-cre元件验证引物p1109(unknown)
<400>19
cggagaatatggagcttcatcgaatcac28
<210>20
<211>28
<212>dna
<213>tet-on-cre元件验证引物p604(unknown)
<400>20
gctctagagctgacgctgacttggatgc28
<210>21
<211>34
<212>dna
<213>c4t318基因扩增引物p1176(unknown)
<400>21
ggaattcatgttcaataacgaacaccacattcca34
<210>22
<211>29
<212>dna
<213>c4t318基因扩增引物p1177(unknown)
<400>22
ggggtaccgggagtaacgccgtgagatgc29
- 还没有人留言评论。精彩留言会获得点赞!