一种用于敲除CCR5基因的gRNA、gRNA组合物和CRISPR-Cas9系统及其用途的制作方法
本发明涉及一种用于敲除ccr5基因的grna、grna组合物和crispr-cas9系统及其用途。
背景技术:
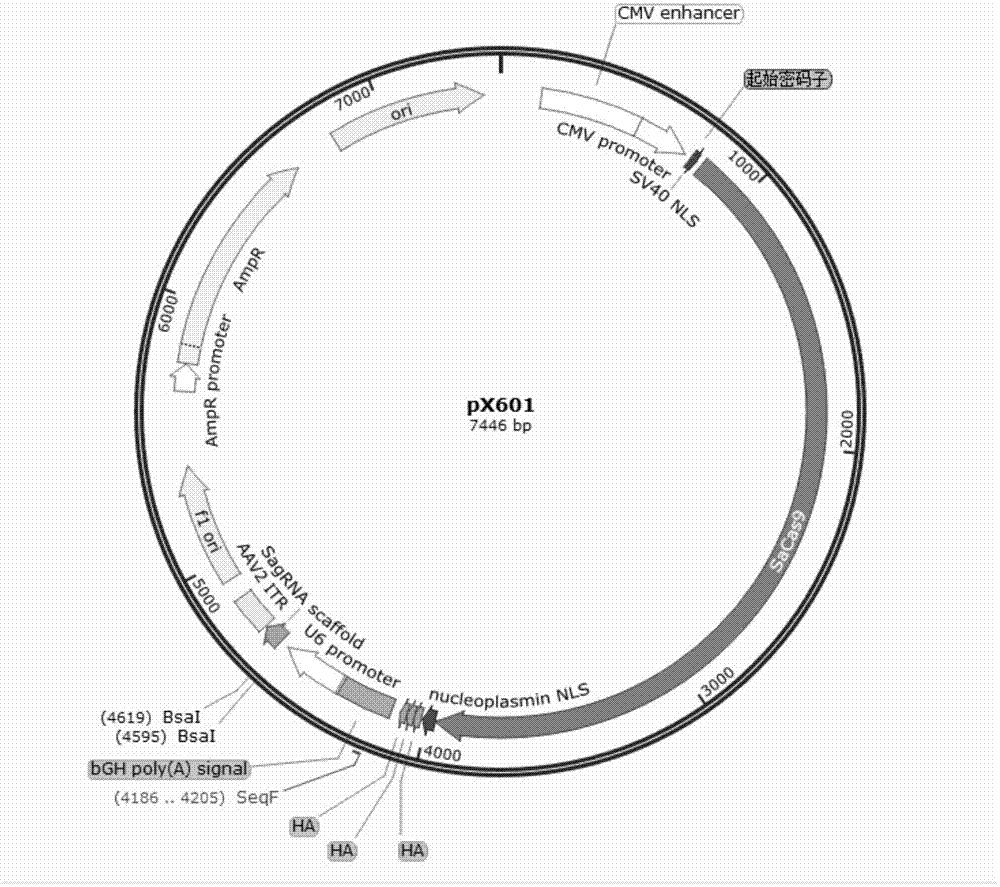
:获得性免疫缺陷综合征(acquiredimmunodeficiencysyndrome,aids)是由人类免疫缺陷病毒(humanimmunodeficiencyvirus,hiv)感染所引起的免疫系统疾病,是一种危害性极大的全球化传染病。hiv以人体免疫系统中最重要的t淋巴细胞作为主要攻击对象,造成免疫功能丧失,从而易患各种疾病,死亡率高达99%-100%。hiv在人体内的潜伏期能长达8-10年,很多hiv感染者在发展成艾滋病患者之前可以无病症的生活,但病毒在人体内始终存在并不可治愈。虽然全世界众多医学研究人员专注于艾滋病的预防和治疗,但至今尚未研制出特效药物,由于hiv的变异极其迅速,因此也还没有可用于预防的有效疫苗。对于hiv感染的治疗,目前的主要策略是最大限度和持久的降低病毒载量、获得免疫功能重建和维持免疫功能、提高生活质量,以及降低hiv相关的发病率和死亡率。抗逆转录病毒治疗(anti-retroviraltherapy,art)是近年来广泛使用的治疗手段,它有效抑制hiv在体内的复制,减少体内病毒载量,从而延长感染者的寿命。但药物副作用、病毒的耐药性突变、终身服药的依从性等都是抗逆转录病毒治疗在临床使用上不可避免的问题。而以基因为基础的治疗方法可以持续的抑制病毒从而减少治疗干预措施,是一种非常有前景的治疗方式。随着基因编辑技术的兴起,多国科学家都尝试通过zfn、talent、crispr-cas9等基因编辑技术,敲除hiv入侵和整合过程中的关键基因,从而干预病毒的转录、复制和扩散。全球首例hiv感染的治愈发生在“柏林病人”身上,他接受了一名ccr5-δ32基因突变纯合体捐献者的骨髓移植,此后的五年都没有在其身上检测到hiv病毒的踪迹。hiv-1主要有r5-嗜性和x4-嗜性两种,对应的辅助受体分别为ccr5和cxcr4,r5-嗜性病毒是目前最普遍感染且在感染的前期起主导作用的病毒类型。通过基因编辑的方式敲除或抑制细胞(如cd4+t细胞、淋巴细胞、骨髓干细胞等)表面的hiv病毒辅助受体ccr5的表达,抑制病毒扩散从而延缓和治疗艾滋病,已经成为hiv基因治疗研究的常见思路。目前世界上公认的三代基因编辑技术为:第一代,zfn;第二代,talen;第三代,crispr。相对于zfn、talen等技术,crispr具有明显的优势,例如构建简单、效率高使用成本低。根据细菌来源不同,目前已经研发出不同类型的crispr酶系统,例如spcas9(来源于streptococcuspyogenes)、sacas9(来源于staphylococcusaureus)、nmcas9(来源于neisseriameningitidis)以及stcas9(来源于streptococcusthermophilus)等系统,但所有的crispr系统均包含以下几个部分,如图1所示:1)pam位点,位于靶序列的下游,只有几个nt(例如,spcas9/ngg;sacas9/nngrrt),根据此位点来选取靶序列;2)sgrna,识别并与切割位点结合的序列,一般在20nt左右;3)tracrrna,连接于sgrna之后的一段回文rna序列;4)cas9内切酶,含有2个独特的活性位点,分别为在氨基末端的ruvc和蛋白质中部的hnh。crispr-cas9基因编辑系统的工作原理为:sgrna、tracrrna和cas9组成复合体,识别并结合于sgrna互补的序列,然后解开dna双链,形成r-loop,使sgrna与互补链杂交,另一条链保持游离的单链状态,然后由cas9中的hnh活性位点剪切sgrna的互补dna链,ruvc活性位点剪切非互补链,最终引入dna双链断裂(dsb)。断裂的dna双链倾向于使用非同源性末端接合(non-homologousendjoining,nhej)的方式进行修复,这种修复不需要任何模版的帮助,因此容易引入突变。这就是利用crispr-cas9系统敲除目的基因的原理。现有技术采用zfn来敲除ccr5基因。zfn由一个dna识别域和一个非特异性核酸内切酶构成。dna识别域一般由3个锌指蛋白串联组成,每个锌指蛋白特异性识别并结合一个三联体碱基,整个识别域共识别9个碱基;非特异性核酸内切酶来自foki的c端96个氨基酸组成的dna剪切域,只在二聚体状态时才有酶切活性。每个foki单体与一个锌指蛋白组相连构成一个zfn,识别特定的位点,当两个识别位点距离恰当时,两个foki单体结合才能产生酶切功能,达到dna定点剪切的目的。zfn由一个dna识别域和一个foki单体组成,且必须二聚化,形成foki二聚体才能产生切割效果。zfn二聚体的两个dna识别域必须识别正义链上9个碱基和反义链上9个碱基,且正反义链的识别区域之间必须间隔适当距离,才能给两个foki单体的结合提供适当的空间。zfn系统虽能保证特异性,但设计和使用较为繁琐和复杂,该识别域为蛋白质,合成费用也较高。技术实现要素:本发明提供了一种用于敲除ccr5基因的grna、grna组合物、crispr-cas9系统和试剂盒及其用途,本发明利用所提供的特异性靶向人ccr5基因的grna序列,结合staphylococcusaureus来源的crispr-cas9系统,解决了ccr5基因靶向突变的问题。ccr5基因的突变,可以抑制病毒扩散从而延缓和治疗艾滋病,可作为艾滋病治疗的新手段。为实现上述目的,所采取的技术方案:一种用于敲除ccr5基因的grna,所述grna的靶序列如seqidno:1~seqidno:25中的一种所示。优选地,所述grna的靶序列如seqidno:3、seqidno:6、seqidno:11、seqidno:12、seqidno:17、seqidno:18、seqidno:19、seqidno:20、seqidno:21中的一种所示。本发明提供了一种用于敲除ccr5基因的grna组合物,所述grna组合物由靶序列如seqidno:11所示的grna和靶序列如seqidno:20所示的grna组成,或由靶序列如seqidno:11所示的grna和靶序列如seqidno:18所示的grna组成,或由靶序列如seqidno:12所示的grna和靶序列如seqidno:20所示的grna组成,或由靶序列如seqidno:17所示的grna和靶序列如seqidno:20所示的grna组成,或由靶序列如seqidno:17所示的grna和靶序列如seqidno:21所示的grna组成,或由靶序列如seqidno:20所示的grna和靶序列如seqidno:21所示的grna组成。本发明提供了一种用于敲除ccr5基因的表达载体,所述表达载体是将上述所述的grna的dna序列或者上述所述的grna组合物的dna序列连接到基础载体上而得的,所述表达载体表达上述所述的grna。优选地,所述基础载体为px601质粒。本发明提供了一种用于敲除ccr5基因的crispr-cas9系统,包括上述所述的grna、或者所述的grna组合物,以及cas9蛋白。优选地,所述cas9蛋白为来源于金黄色酿脓葡萄球菌staphylococcusaureus的cas9蛋白。本发明提供了一种用于敲除ccr5基因的试剂盒,其特征在于,包括:(1)表达cas9蛋白的载体、cas9蛋白和cas9蛋白对应的mrna中的至少一种;(2)上述所述的grna和表达上述所述的grna的载体中的至少一种、或者上述所述的grna组合物和表达上述所述的grna组合物的载体中的至少一种。本发明提供了一种重组杆状病毒,所述重组杆状病毒是将如权利要求3所述的grna组合物的dna序列和staphylococcusaureus来源的cas9蛋白的dna序列连接到pfast-bac表达载体上后包装到杆状病毒而得的,所述重组杆状病毒表达所述grna组合物和所述cas9蛋白。本发明提供了上述所述的grna、上述所述的grna组合物、上述所述的表达载体或者上述所述的crispr-cas9系统在制备预防或治疗hiv感染的药物中的用途。本发明提供了上述所述的重组杆状病毒在制备用于编辑哺乳动物细胞中ccr5基因的试剂中的用途。本发明的有益效果在于:本发明使用crispr-cas9技术,对hiv病毒进入人体细胞的辅助受体ccr5基因进行切割,设计和操作简便、突变效率高,成本低,而且有效实现ccr5基因的突变,可用于艾滋病基因治疗技术的开发。附图说明图1为staphylococcusaureus来源crispr-cas9载体px601;图2为staphylococcusaureus来源crispr-cas9系统对ccr5基因的突变效率检测;图3为staphylococcusaureus来源crispr-cas9系统对ccr5基因突变效率和突变类型分析;图4为staphylococcusaureus来源crispr-cas9系统对ccr5蛋白敲除效率分析;图5为实施例4中crispr-cas9联合grna组合物对ccr5基因进行t7e1酶切检测的电泳图;图6为实施例5中12+20对应的随机单菌落sanger测序结果分析;图7为实施例5中17+21对应的随机单菌落sanger测序结果分析;图8为实施例5中17+20对应的随机单菌落sanger测序结果分析;图9为实施例5中20+21对应的随机单菌落sanger测序结果分析;图10为实施例6中电转染后cd4+t细胞的ccr5基因突变效率检测;图11为实施例6中ccr5基因突变cd4+t细胞对hiv病毒的拮抗作用;图12为实施例7中pfast-ef1α-egfp-cmv-sacas9质粒图谱;图13为实施例7中pfast-r20-egfp-sacas9-f12载体图谱;图14为实施例7中杆状病毒感染hela细胞的ccr5基因突变效率检测。具体实施方式为更好的说明本发明的目的、技术方案和优点,下面将结合具体实施例对本发明作进一步说明。实施例1采用crispr-cas9技术突变ccr5基因1.1grna准备(1)根据ccr5基因的序列设计21nt的grna序列,所述grna的靶序列如seqidno:1-seqidno:25中的一种所示;(2)分别合成grna对应的dna序列正义链和反义链(正义链的5’-端加cacc,若正义链5’-端第一个核苷酸不是鸟嘌呤g,则在正义链的5’-端加caccg;在反义链的5’-端加aaac,若正义链5’-端第一个核苷酸不是鸟嘌呤g,则在反义链的3’-端加c);(3)将上述grna正义链和反义链混合,90℃处理后自然冷却至室温进行退火处理,合成带粘性末端的双链dna,所述双链dna用于转录出相应的grna。针对ccr5基因所设计的grna对应的dna序列正义链如表1所示:表1针对ccr5基因所设计的grna对应的正义链(或靶序列)1.2载体准备(1)px601质粒(图1)扩增和提取,并测定质粒浓度;(2)采用限制性内切酶bsai对px601进行酶切,37℃酶切1h后加loadingbuffer终止反应。(3)琼脂糖凝胶电泳后切胶回收线性化质粒px601,并测定回收产物浓度,-20℃保存备用。1.3连接转化(1)将切胶回收的线性化px601载体与退火后的双链dna进行连接反应;(2)连接产物热击法转化大肠杆菌感受态细胞top10,转化后向每个离心管加入无菌的lb液体培养基(不含抗生素),混匀后置于恒温摇床37℃200rpm振荡培养45min使菌体复苏。(3)将复苏后的top10细胞涂布lb固体平板(amp+),倒置于恒温培养箱37℃静置培养12-16h。(4)从上述平板上分别挑取单菌落接种到lb液体培养基(amp+)中扩大培养。(5)使用引物seqf(5’-ttccttgaccctggaaggtg-3’)(seqidno:26),对上述菌液分别测序;(6)将测序正确的菌液提取质粒并测定质粒浓度后至-20℃保存备用。1.4细胞转染(1)hek293t细胞或hela细胞铺板;(2)采用lipofectamine3000试剂盒将1.3(6)中提取的质粒转染hek293t细胞或者hela细胞;特别注意的是,hek293t细胞ccr5基因的其中一个等位基因在δ32区存在突变,故对δ32区内的grna的突变效率分析不宜采用hek293t细胞,本方案中,我们对δ32区内的grna的突变效率分析使用hela细胞。转染方法同hek293t。(3)转染后的细胞培养48小时,离心收集细胞。1.5t7e1酶切分析突变效率(1)将上述1.4(3)收集的细胞提取细胞基因组,并检测基因组浓度;(2)分别在grna结合位点上下游设计pcr引物,如表2所示;表2t7e1酶切分析引物列表(3)分别使用pcr法扩增带有靶位点的目的片段;(4)纯化回收pcr产物,并测定产物浓度;(5)将上述纯化的pcr产物退火处理,即先加热至95℃,保温10min,然后以每30s下降2~3℃的速度降温至室温;(6)上述每管退火产物分别加入t7核酸内切酶1(t7e1),设置mock组(未转化的细胞)和空白对照(ck)组(不加t7e1,用ddh2o代替),37℃酶切1h。(7)2%琼脂糖凝胶电泳检测酶切效果,marker为购于北京康为世纪生物科技有限公司的cw0636s,各条带大小从上至下分别为:1500bp、1000bp、800bp、600bp、500bp、400bp、300bp、200bp、100bp,见图2。结果如图2所示,(a)为t1区的检测结果,(b)为δ32区的检测结果(c)为t2区的检测结果。电泳图中各泳道编号分别对应各grna的dna序列编号,即泳道1-25对应seqidno:1-seqidno:25。其中泳道3、6、11、12、17、18、19、20、21与mock和ck相比,具有明显的切割条带。前述grna与ccr5基因的结合位点离两端引物的距离和被切割的条带大小相吻合,且mock和ck组没有切割条带,说明seqidno:3、seqidno:6、seqidno:11、seqidno:12、seqidno:17、seqidno:18、seqidno:19、seqidno:20、seqidno:21所对应的grna能引导cas9蛋白对ccr5基因进行有效切割。实施例2测序分析grna对ccr5基因突变效率和突变类型在实施例1筛选出来的切割效果较优的grna中(seqidno:3、seqidno:6、seqidno:11、seqidno:12、seqidno:17、seqidno:18、seqidno:19、seqidno:20、seqidno:21),随机选取一个grna,采用crispr-cas9基因编辑系统,靶向突变ccr5基因,并用sanger测序的方法进一步确认对ccr5基因的突变效率和突变类型。以下操作以seqidno:12为例进行。2.1将实施例1中1.5(4)所得的纯化后的pcr产物,测定浓度备用;2.2选用康为世纪masterpcrmix给pcr产物3’-末端加一个腺嘌呤(a):取2.1中的pcr产物2μg按照1:1(v:v)比例与masterpcrmix混合,72℃反应30min;2.3用1%琼脂糖凝胶电泳2.2中的反应产物,切胶回收目的片段并测定回收产物浓度;2.4用takarapmdtm18-tvectorcloningkit进行连接反应:按下表配制反应体系,16℃反应30min;表3连接pmdtm18-t载体的反应体系ddh2o补齐至10μlpmd18-tvector(5×)1μl(10ng)目的片段0.1pmol~0.3pmolsolutioni5μl2.5上述连接产物热击法转化大肠杆菌感受态细胞top10,转化后向每个离心管加入无菌的lb液体培养基(不含抗生素),混匀后置于恒温摇床37℃200rpm振荡培养45min使菌体复苏;2.6将复苏后的top10细胞涂布lb固体平板(amp+),倒置于恒温培养箱37℃静置培养12-16h;2.7随机挑取平板上长出来的单菌落,进行sanger测序,结果如下:共对54个随机挑选的单菌落测序,其中发生突变的18个,突变效率为33.3%,突变类型如图3所示:左侧第一栏数字代表随机挑选的菌落编号,wildtype为未经基因编辑处理的原始ccr5基因序列,******表示grna识别靶定基因所必需的pam区,灰色区域为grna识别的靶序列,,-表示碱基缺失。由图3可见,共有11个单菌落(4、8、10、13、17、23、25、27、29、41、64号菌)为缺失突变,5个单菌落为插入突变(5、15、37、56、65号菌)为插入突变,1个单菌落为多点突变(19号),1个单菌落为大片段缺失(9号)。整体突变效率为33.3%。实施例3westernblot分析ccr5蛋白敲除效率在实施例1筛选出来的切割效果较优的grna中(seqidno:3、seqidno:6、seqidno:11、seqidno:12、seqidno:17、seqidno:18、seqidno:19、seqidno:20、seqidno:21),随机选取1个grna,采用crispr-cas9基因编辑系统,靶向突变ccr5基因,用westernblot进一步分析对ccr5蛋白的敲除效率。以下以seqidno:20为例进行操作和分析。3.1总蛋白抽提(1)按1ml裂解液加10μlpmsf(100mm)和10μlcocktail,摇匀置于冰上(pmsf要摇匀至无结晶时才可与裂解液混合)。(2)每管细胞加100μl含pmsf和的裂解液,于冰上裂解30min,为使细胞充分裂解要经常来回摇动。(3)裂解完后于4℃下14000rpm离心5min。(提前开离心机预冷)。(4)将离心后的上清分装转移倒干净的离心管中放于-80℃保存备用。3.2蛋白样品初步定量采用蛋白质定量的经典方法bca法,测定上述抽提的各组细胞样品的蛋白质浓度。将各组样品的蛋白质浓度调整成一致。将调整好的样品和5×上样缓冲液按4:1混合,煮沸10min,缓慢恢复室温后,稍离心,放于-20℃保存备用。3.3sds-page电泳,各样品上样体积保持一致,分离胶浓度10%,80v恒压电泳至溴酚蓝进入分离胶后,120v恒压电泳至溴酚蓝刚出胶底部停止电泳。3.4蛋白质转移(1)pvdf膜甲醇预处理3~5秒,放至转膜缓冲液浸润半小时。(2)取出凝胶,将其放至滤纸上,形成凝胶转印堆积层,滤纸,凝胶,pvdf膜,滤纸,凝胶转印堆积层这样的“三明治”结构。此操作必须将气泡完全去除。(3)按正负极方向放置转印夹。(4)低温条件下,100v恒压根据蛋白分子量每1kda转膜1分钟来进行转膜。3.5免疫印记(1)取出杂交膜,tbst漂洗5min,1次。(2)5%脱脂奶粉溶液室温封闭1小时。(3)tbst洗膜10min,1次。(4)用一抗稀释液,在4℃孵育过夜(所用的一抗为proteintech公司生产的ccr5抗体,稀释倍数1:1000)。(5)tbst洗膜10min,4次。(6)用二抗稀释液37℃孵育1h(所用的二抗为southernbiotech公司生产的goatanti-rabbitigg(h+l),mouse/humanads-hrp,稀释倍数1:20000)。(7)tbst洗膜10min,4次。(8)将杂交膜置于一透明塑料板上,注意不要让膜干燥。(9)用一干净移液器将化学荧光发光底物均匀地加到膜的表面,并使反应持续5min。(10)用试剂盒提供的滤纸吸去膜表面多余的底物溶液,放至暗盒,显影。3.6杂交结果见图4,灰度分析结果见表4。其中以gapdh基因作为内参,样品编号与grna对应的靶序列编号一致。表4crispr-cas9对ccr5基因的敲除效率分析样品编号mockseqidno:20ccr5灰度值847.13601.72gapdh灰度值1811.831925.56ccr5敲除效率——33.16%实施例4crispr-cas9联合grna组合物突变ccr5基因4.1将实施例1中1.3(6)制备的连接有seqidno:11、seqidno:12、seqidno:17、seqidno:18、seqidno:20、seqidno:21的px601载体,进行如下组合:px601-seqidno:11和px601-seqidno:20、px601-seqidno:11和px601-seqidno:18、px601-seqidno:12和px601-seqidno:20、px601-seqidno:17和px601-seqidno:20、px601-seqidno:17和px601-seqidno:21、px601-seqidno:20和px601-seqidno:21。4.2将上述6个组合按照1.4的方法分别转化hela细胞,再按照1.5的步骤进行t7e1酶切分析切割效率,结果如图5所示:(a)、(b)为在t7e1酶切前对转染组和对照组mock(没有转染的hela细胞)的含有靶序列的片段进行pcr的结果,泳道“11+20”、“11+18”、“12+20”、“17+21”、“17+20”、“20+21”分别表示seqidno:11和seqidno:20、seqidno:11和seqidno:18、seqidno:12和seqidno:20、seqidno:17和seqidno:21、seqidno:17和seqidno:20、seqidno:20和seqidno:21以1:1的摩尔比混合共转hela细胞细胞。由于seqidno:11和seqidno:20、seqidno:11和seqidno:18、seqidno:12和seqidno:20、seqidno:17和seqidno:21、seqidno:17和seqidno:20对应的靶序列在ccr5基因上距离较大,两个guiderna共转染会造成大片段缺失,因此在电泳图上可见转染组pcr结果呈现两个条带,其中分子量较大的条带与mock组pcr条带迁移率相同,判断为未发生大片段缺失的pcr产物;而转染组分子量较小的pcr条带与mock组的pcr条带相比略小,判断为发生大片段缺失的pcr产物。图5中(c)为对照组mock和转染组(seqidno:20和seqidno:21)的t7e1酶切结果电泳图。根据pcr和t7e1酶切的结果,六种双grna联合使用均能对ccr5基因进行有效切割。实施例5测序分析grna组合物对ccr5基因突变效率和突变类型选取实施例4中的seqidno:12和seqidno:20、seqidno:17和seqidno:21、seqidno:17和seqidno:20、seqidno:20和seqidno:21的grna组合物,采用crispr-cas9基因编辑系统,靶向突变ccr5基因,并用sanger测序的方法进一步确认对ccr5基因的突变效率和突变类型。5.1按照实施例2中2.1~2.7步骤制备平板,随机挑选平板上的单菌落进行sanger测序,结果如下:12+20对应的单菌落随机挑选55个进行sanger测序,载体自连的假阳性单菌落为4个,阳性单菌落51个,其中发生突变的33个,突变效率为64.7%,突变类型图6所示。菌落1、2、5、13、16、21、23、25、28、31、32、34、41、42、43、46、48、49、50在靶位点seqidno:12和seqidno:20之间发生227bp的长片段缺失,且菌落50有1bp替换;菌落18、20、27、36、38、45在靶位点seqidno:12和seqidno:20之间发生了207bp的长片段颠倒,且在pam序列两端有碱基插入;菌落3在靶位点seqidno:12处有碱基插入,在靶位点seqidno:20处有碱基缺失和插入;菌落29在靶位点seqidno:20处有大片段插入;菌落30在靶位点seqidno:20处有碱基替换;菌落40在靶位点seqidno:12和seqidno:12处均有碱基缺失;菌落35在靶位点seqidno:20前发生大片段缺失;菌落39、51突变序列混乱。17+21对对应的单菌落随机挑选20个进行sanger测序,载体自连的假阳性单菌落为2个,阳性单菌落18个,其中发生突变的6个,突变效率为31.6%,突变类型图7所示。菌落3、5、14、19、16在靶位点seqidno:17和seqidno:21之间发生185~192bp的长片段缺失;菌落8在靶位点seqidno:17处发生了1bp插入。17+20对对应的单菌落随机挑选55个进行sanger测序,载体自连的假阳性单菌落为5个,阳性单菌落50个,其中发生突变的21个,突变效率为42%,突变类型图8所示。菌落2、4、10、11、14、23、26、28、31、40、42、48、49、50、55、13、37在靶位点seqidno:17和seqidno:20之间发生128~131bp的长片段缺失;菌落30在靶位点seqidno:17后发生大片段缺失;菌落47在靶位点seqidno:20处发生了75bp缺失;菌落27在靶位点seqidno:17和seqidno:20处均有碱基缺失;菌落9在靶位点seqidno:17处有碱基替换。20+21对应的单菌落随机挑选29个进行sanger测序,载体自连的假阳性单菌落为3个,阳性单菌落26个,其中发生突变的7个,突变效率为26.9%,突变类型图9所示。菌落1、8、26在靶位点seqidno:20和seqidno:21之间发生57bp的长片段缺失,且在靶位点seqidno:21处有碱基替换;菌落5、11在靶位点seqidno:21处有碱基替换;菌落10在靶位点seqidno:20处有碱基替换;菌落27在靶位点seqidno:20处有碱基缺失。上述结果表明,px601-seqidno:12和px601-seqidno:20、px601-seqidno:17和px601-seqidno:21、px601-seqidno:17和px601-seqidno:20、px601-seqidno:20和px601-seqidno:21共同转化hela细胞,都能对ccr5基因进行有效突变,其中seqidno:12和seqidno:20双grna联合使用切割效率明显高于其他3个双grna联合使用。实施例6ccr5基因突变后t细胞对hiv病毒的拮抗作用6.1cd4+t细胞ccr5基因突变6.1.1质粒准备:将实施例1中的质粒px601-seqidno:12和px601-seqidno:20扩增和提取,并测定质粒浓度;6.1.2细胞电转染:(1)收取cd4+t细胞进行细胞计数。(2)solutioua配制:将16.4μlp3primarycellsolution与3.6μlsupplement1混匀,静置备用。(3)solutioub配制:取4μlsolutioua,加入1.5μg质粒px601-seqidno:12和1.5μg质粒px601-seqidno:20,充分混匀。(4)取每孔5.5×105cell于1.5mlrnase-free离心管中,100g离心10min后,充分去除培养基,加入16μlsolutioua混匀细胞。(5)将solutioub与(4)中的细胞悬液混匀。加入到16wellnucleofectortmstrip,选择电转程序,将nucleofectortmstrip放置电转仪内,开始电转。(6)电转结束后加入100μl的cd4+t细胞培养基于nucleofectortmstrip,轻轻混匀细胞,转移到48孔板内,放入37℃、5%co2和95%相对湿度的培养箱中,进行培养。(7)电转染后的细胞培养48h,离心收集细胞。(8)按照1.5的步骤进行t7e1酶切分析切割效率,结果如图10所示,对转染组和对照组ck(没有电转染的cd4-t细胞细胞)的含有靶序列的片段进行pcr的结果,泳道“12+20”表示电转质粒px601-seqidno:12和px601-seqidno:20。由于seqidno:12和px601-seqidno:20对应的靶序列在ccr5基因序列上距离为227bp,两个质粒共转染会造成大片段缺失,因此在电泳图上可见转染组pcr结果呈现两个条带,其中分子量较大的条带与ck组pcr条带迁移率相同,判断为未发生大片段缺失的pcr产物;而转染组分子量较小的pcr条带与mock组的pcr条带相比略小,判断为发生大片段缺失的pcr产物。6.2hiv-1r5攻毒实验(1)将本实施例6.1中获得的ccr5基因突变后cd4+t细胞,在培养基中加入5μg/mlpha-l培养3d后,铺96孔板,铺板密度为1.0×105cells/孔,用rpmimedium1640basic培养基(含10%fbs+500iu/mlil-2)培养过夜,以未改造的cd4+t细胞为对照组,每组设3个重复;(2)向培养基中添加hiv-1r5(bal),病毒含量为12ng/孔,侵染过夜;(3)弃含病毒的培养上清液,换300μl新鲜的rpmimedium1640basic培养基(含10%fbs+500iu/mlil-2);(4)病毒侵染的第4天,取150μl上清液放-80℃保存待测,添加150μlrpmimedium1640basic培养基(含10%fbs+500iu/mlil-2);(5)病毒侵染的第7天,取150μl上清液放-80℃保存待测。(6)使用hiv-1p24抗原检测试剂盒对第4天和第7天的样品进行检测,结果如图11所示:被hiv-1r5(bal)侵染的第4天,对照组中检测到256002.28pg/ml的p24蛋白,而ccr5基因突变cd4+t细胞在第4天检测到了98484.28pg/ml的p24蛋白,明显低于对照组;在第7天时,对照组中p24蛋白达384940.04pg/ml,即病毒在对照组细胞中随时间大量扩增繁殖,而ccr5基因突变cd4+t细胞中检测到了147612.92pg/ml的p24蛋白,病毒在ccr5基因突变cd4+t细胞中扩增缓慢。说明ccr5基因突变cd4+t细胞对hiv-1r5(bal)的侵染和扩散有抵抗作用。实施例7运载crispr-cas9的杆状病毒包装及细胞感染7.1pfast-egfp-sacas9-f12载体构建7.1.1载体准备(1)pfast-ef1α-egfp-cmv-sacas9质粒(图12)扩增和提取,并测定质粒浓度;(2)采用限制性内切酶bstbi对pfast-ef1α-egfp-cmv-sacas9进行酶切,65℃酶切1h后加loadingbuffer终止反应。(3)琼脂糖凝胶电泳后切胶回收线性化质粒pfast-ef1α-egfp-cmv-sacas9,并测定回收产物浓度,-20℃保存备用。7.1.2pcr扩增目的片段u6-seqidno:12-grna(1)以px601-seqidno:12为模板,引物为seqidno:33和seqidno:34为引物扩增目的片段u6-seqidno:12-grna,pcr产物用1%琼脂糖凝胶电泳检测后切胶回收目的片段;表5目的片段u6-seqidno:12-grna扩增引物33、347.1.3重组反应酶切后的pfast-ef1α-egfp-cmv-sacas9和目的片段u6-seqidno:12-grna进行同源重组,37℃重组30min,冰浴5min;7.1.4转化(1)按照实施例1中1.3(2)~(4)步骤,将重组连接产物转化大肠杆菌感受态细胞dh5α,随后从平板上分别挑取单菌落接种到lb液体培养基(amp+)中扩大培养,并对上述菌液测序。(2)将测序正确的菌液提取质粒并测定质粒浓度后至-20℃保存备用。7.2pfast-r20-egfp-sacas9-f12载体构建7.2.1pcr扩增目的片段u6-seqidno:20-grna(1)以px601-seqidno:20为模板,引物为seqidno:35和seqidno:36为引物扩增目的片段u6-seqidno:20-grna,pcr产物用1%琼脂糖凝胶电泳检测后切胶回收目的片段;表6目的片段u6-seqidno:20-grna扩增引物35、367.2.2重组反应(1)将本实施例1.1中pfast-egfp-sacas9-f12质粒,用bsu36i,37℃酶切1h后,80℃失活20min;(2)上述酶切产物与目的片段u6-seqidno:20-grna进行同源重组,37℃重组30min,冰浴5min;7.2.3转化(1)按照实施例1中1.3(2)~(4)步骤,将重组连接产物转化大肠杆菌感受态细胞dh5α,随后从平板上分别挑取单菌落接种到lb液体培养基(amp+)中扩大培养,并对上述菌液测序,(2)将测序正确的菌液提取质粒(如图13)并测定质粒浓度后至-20℃保存备用。7.3杆粒dna制备7.3.1转化感受态dh10bac(1)dh10bac感受态细胞加入本实施例7.2中制备的pfast-r20-egfp-sacas9-f12质粒,混匀,冰上静置30min;(2)42℃金属浴热激45s,冰浴静置5min;(3)向离心管中加入450μllb培养基(无抗生素),摇床37℃,200rpm培养5h;(4涂lb固体平板(含25μg/mlkan,3.5μg/mlgen,5μg/mltet,50μg/mlx-gal,20μg/mliptg);(5)培养箱37℃培养48h,观察是否有白色克隆。7.3.2提取重组杆粒dna(1)用划线棒挑取白色克隆接种到lb液体培养基(含50μg/mlkan,1.4μg/mlgen,10μg/mltet),恒温摇床37℃,200rpm培养22h;(2)提取杆粒dna并将dna储存在4℃备用。7.4杆状病毒包装7.4.1重组杆粒转染sf9细胞及p1病毒收取(1)取9×105个sf9细胞接种于细胞培养板,培养箱27℃孵育30min~1h;(2)试剂配制:solutiona:8μlcellfectiniireagent+100μlgrace’smedium,unsupplemented,移液枪轻轻混匀,室温静置5min;solutionb:3μgbacmid+100μlgrace’smedium,unsupplemented,移液枪轻轻混匀,室温静置5min;dna-lipidmixture:将solutiona与solutionb混合成dna-lipidmixture,室温孵育30min;(3)弃去细胞培养液,将200μldna-lipidmixture加入细胞培养板中,轻轻摇动混匀;(4)培养箱27℃孵育5h;(5)去除dna-lipidmixture,添加2ml新鲜sf-900iisfm培养基,27℃孵育72h;(6)收集培养液上清,4℃、1000g离心10min,取上清,4℃保存备用。7.4.2p2病毒获得(1)在t75培养瓶中接种5mlsf9细胞悬液,在27℃培养箱孵育1h;(2)加入p1病毒液,27℃培养箱过夜;(3)补加5ml室温预热的sf-900iisfm培养基;(4)27℃培养箱孵育3~4d;(5)收集培养液上清,4℃、1000g离心10min,取上清,4℃保存备用。7.4.3p3病毒获得(1)在t175培养瓶中接种12mlsf9细胞悬液,在27℃培养箱孵育1h;(2)加入p1病毒液,27℃培养箱过夜;(3)补加10ml室温预热的sf-900iisfm培养基;(4)27℃培养箱孵育3~4d;(5)收集培养液上清,4℃、1000g离心10min,取上清;(6)上述上清液于4℃,28000g离心90min,弃上清,获得p3病毒,4℃储存备用。7.5杆状病毒感染hela细胞(1)hela细胞铺板,37℃、5%co2培养24h;(2)把原培养基移除,并用pbs洗涤细胞一次,加入杆状病毒与不含血清的dmembasic培养基,37℃、5%co2培养,24h后换液;(3)病毒感染72h后,离心收集细胞。按照实施例1中1.5的步骤进行t7e1酶切分析切割效率。如图14所示为对感染组(泳道“12+20”)和对照组ck(没有感染病毒的hela细胞)的含有靶序列的片段进行pcr的结果。由于运载crispr-cas9的杆状病毒携带seqidno:12和seqidno:20,seqidno:12和seqidno:20对应的靶序列在ccr5基因序列上距离为227bp,病毒感染导致其所携带的crispr-cas9系统在细胞基因组上进行切割,因此在电泳图上可见感染组pcr结果呈现两个条带,其中分子量较大的条带与ck组pcr条带迁移率相同,判断为未发生大片段缺失的pcr产物;而感染组分子量较小的pcr条带与ck组的pcr条带相比略小,判断为发生大片段缺失的pcr产物。最后所应当说明的是,以上实施例仅用以说明本发明的技术方案而非对本发明保护范围的限制,尽管参照较佳实施例对本发明作了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的实质和范围。序列表<110>广东赤萌医疗科技有限公司<120>一种用于敲除ccr5基因的grna、grna组合物和crispr-cas9系统及其用途<130>2018<160>36<170>patentinversion3.3<210>1<211>21<212>dna<213>人工序列<400>1agatcactttttatttatgca21<210>2<211>21<212>dna<213>人工序列<400>2ttatgcacagggtggaacaag21<210>3<211>21<212>dna<213>人工序列<400>3caaaaccaaagatgaacacca21<210>4<211>21<212>dna<213>人工序列<400>4ccttttgcagtttatcaggat21<210>5<211>21<212>dna<213>人工序列<400>5cttcagccttttgcagtttat21<210>6<211>21<212>dna<213>人工序列<400>6cctataaaatagagccctgtc21<210>7<211>21<212>dna<213>人工序列<400>7ctattttataggcttcttctc21<210>8<211>21<212>dna<213>人工序列<400>8caggtacctatcgattgtcag21<210>9<211>21<212>dna<213>人工序列<400>9aaaagccaggacggtcacctt21<210>10<211>21<212>dna<213>人工序列<400>10ggtggtgacaagtgtgatcac21<210>11<211>21<212>dna<213>人工序列<400>11ggctgtgtttgcgtctctccc21<210>12<211>21<212>dna<213>人工序列<400>12tacagtcagtatcaattctgg21<210>13<211>21<212>dna<213>人工序列<400>13tttaatgtctggaaattcttc21<210>14<211>21<212>dna<213>人工序列<400>14tgtcatggtcatctgctactc21<210>15<211>21<212>dna<213>人工序列<400>15gaagcagagtttttaggattc21<210>16<211>21<212>dna<213>人工序列<400>16tcgacaccgaagcagagtttt21<210>17<211>21<212>dna<213>人工序列<400>17cttctcatttcgacaccgaag21<210>18<211>21<212>dna<213>人工序列<400>18tccttctcctgaacaccttcc21<210>19<211>21<212>dna<213>人工序列<400>19accttccaggaattctttggc21<210>20<211>21<212>dna<213>人工序列<400>20ctactgcaattattcaggcca21<210>21<211>21<212>dna<213>人工序列<400>21tatgcaggtgacagagactct21<210>22<211>21<212>dna<213>人工序列<400>22gatgcagcagtgcgtcatccc21<210>23<211>21<212>dna<213>人工序列<400>23ccccgacaaaggcatagatga21<210>24<211>21<212>dna<213>人工序列<400>24tatttcctgctccccagtgga21<210>25<211>21<212>dna<213>人工序列<400>25acagatatttcctgctcccca21<210>26<211>20<212>dna<213>人工序列<400>26ttccttgaccctggaaggtg20<210>27<211>20<212>dna<213>人工序列<400>27catggagggcaactaaatac20<210>28<211>18<212>dna<213>人工序列<400>28agagctgcaggtgtaatg18<210>29<211>22<212>dna<213>人工序列<400>29gagcatgactgacatctacctg22<210>30<211>20<212>dna<213>人工序列<400>30cagaagcgtttggcaatgtg20<210>31<211>19<212>dna<213>人工序列<400>31tgcttgtcatggtcatctg19<210>32<211>20<212>dna<213>人工序列<400>32tggatgaatcttagaccctc20<210>33<211>41<212>dna<213>人工序列<400>33tggggaggtaccgattcgaagaggtaccgagggcctatttc41<210>34<211>44<212>dna<213>人工序列<400>34tgatgacgtcagcgttcgaacaaaaatctcgccaacaagttgac44<210>35<211>41<212>dna<213>人工序列<400>35aggaaggaggaggcctaagggaggtaccgagggcctatttc41<210>36<211>44<212>dna<213>人工序列<400>36agaaaagccccatccttaggcaaaaatctcgccaacaagttgac44当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1