三环类化合物、其制备方法及用途与流程
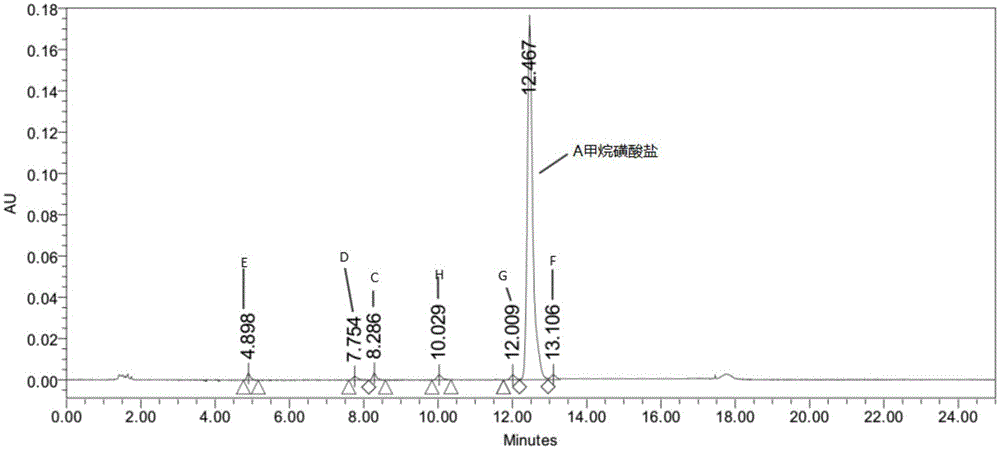
本发明属于医药化学领域,具体涉及三环类化合物、其制备方法及其作为参比对照品用于n-(2-((2-(二甲基氨基)乙基)(甲基)氨基)-4-甲氧基-5-((4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)氨基)苯基)烯丙酰胺甲烷磺酸盐原料药质量研究中相关杂质定性和/或定量分析的用途。
背景技术:
:表皮生长因子受体(egfr)是erbb受体家族的跨膜蛋白酪氨酸激酶成员。erbb受体的同源二聚和/或异源二聚导致胞内域中关键酪氨酸残基的磷酸化,刺激参与细胞增殖和生存的许多细胞内信号传导通路。erbb家族信号传导的失调促进增殖、侵入、转移、血管生成和肿瘤细胞生存,并且已在许多癌症,例如肺癌、头颈部癌和乳腺癌等中得到描述。小分子egfr酪氨酸激酶抑制剂与atp竞争结合到egfr胞内区磷酸化位点,抑制egfr自磷酸过程并阻断下游信号通路,从而达到抑制肿瘤细胞的目的。吉非替尼、厄洛替尼是egfr的第一代可逆小分子抑制剂,主要用于治疗非小细胞肺癌。但是临床研究表明许多患者很快(12-14个月)就对这些egfr小分子抑制剂产生抗药性。研究表明看门残基t790m突变是egfr基因20号外显子一个突变点,是导致耐药产生的主要机制之一。第二代不可逆抑制剂,如阿法替尼对l858r以及t790m突变的egfr具有很强的抑制作用,对吉非替尼或厄洛替尼已经产生抗药性的患者有显著的疗效。然而第二代egfr突变体抑制剂对野生型egfr也同样具有很强的抑制作用,从而导致临床治疗过程中大部分患者产生毒副作用,譬如皮疹、腹泻。因此第三代egfr抑制剂首先应保持对egfrl858r激活突变体、exon19缺失激活突变体以及t790m抗性突变体有较强的抑制作用,同时应克服第二代抑制剂的毒副作用,即减少对野生型egfr的抑制作用。阿斯利康公司(astrazeneca)研发的化合物azd9291(又名n-(2-{2-二甲氨基乙基-甲氨基}-4-甲氧基-5-{[4-(1-甲基吲哚-3-基)嘧啶-2-基]氨基}苯基)丙-2-烯酰胺)是一种口服的、不可逆的egfr抑制剂,该药对egfr-t790m突变阳性非小细胞肺癌患者有较佳的治疗效果。但是它的代谢物az5104对野生型egfr也有很强的抑制作用。因此,仍需开发具有更好药效的新型egfr抑制剂。本发明的发明人研究发现式a的化合物是一种不可逆的新型egfr激酶抑制剂。式a化合物的化学名称为n-(2-((2-(二甲基氨基)乙基)(甲基)氨基)-4-甲氧基-5-((4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)氨基)苯基)烯丙酰胺。体外激酶活性检测发现式a化合物对突变型egfr激酶,例如egfrl858r/t790m激酶具有良好的抑制活性,ic50值小于1nm,对野生型egfr激酶影响小,具有较好的选择性。体外细胞实验结果表明式a的化合物对双突变型细胞的抑制作用较好,且对egfr野生型细胞的抑制小,选择性好。本发明的发明人进一步研究发现式a化合物甲烷磺酸盐水溶性好、生物利用度高,这将有助于提高临床疗效以及降低不良反应等。众所周知,对于人类用药,出于安全性因素要求,国内和国际管理机构对原料药(api)中未确证或毒性未定杂质的限度规定非常低,通常低于0.1%(重量)。需要对这些重要杂质进行深入研究,以确保制备得到符合药用标准、能够用于制备安全有效的药物制剂的原料药。原料药中的杂质可能是由于自身的降解而产生的,也可能来源于制备方法,例如,包括未反应的起始原料、起始原料中包含的杂质的化学衍生物、合成副产物、以及降解产物等。通过了解杂质的化学结构和合成途径,以及通过鉴别影响终产物中杂质含量的参数,能够大大增强对相关杂质的控制。n-(2-((2-(二甲基氨基)乙基)(甲基)氨基)-4-甲氧基-5-((4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)氨基)苯基)烯丙酰胺甲烷磺酸盐作为原料药可能包含多种来源的杂质,对其成药性、安全性和有效性带来隐患。因此,研究其副产物或杂质的性质,对这些副产物或杂质进行检测控制具有重大的意义。技术实现要素:本发明的第一个目的是提供式b、式c、式d、式e、式f、式g、式h或式i所示的化合物或其水合物、溶剂合物或结晶:优选地,分离制备的式b、式c、式d、式e、式f、式g、式h、式i化合物或其水合物、溶剂合物或结晶各包含小于1%的式a化合物(根据hplc判断,面积归一化法),或者分离制备的式b、式c、式d、式e、式f、式g、式h、式i化合物或其水合物、溶剂合物或结晶的纯度至少为85%(根据hplc判断,面积归一化法)。在一些实施方案中,分离制备的式b、式c、式d、式e、式f、式g、式h、式i化合物或其水合物、溶剂合物或结晶的纯度至少为90%(根据hplc判断,面积归一化法)。在另一些实施方案中,分离制备的式b、式c、式d、式e、式f、式g、式h、式i化合物或其水合物、溶剂合物或结晶的纯度至少为95%(根据hplc判断,面积归一化法)。在另一些实施方案中,分离制备的式b、式c、式d、式e、式f、式g、式h、式i化合物或其水合物、溶剂合物或结晶的纯度至少为98%(根据hplc判断,面积归一化法)。在另一些实施方案中,分离制备的式b、式c、式d、式e、式f、式g、式h、式i化合物或其水合物、溶剂合物或结晶的纯度至少为99%(根据hplc判断,面积归一化法)。本发明的第二个目的是提供式b、式c、式d、式e、式f、式g、式h和式i化合物、或其水合物、溶剂合物或结晶的制备方法:所述的式b化合物或其水合物、溶剂合物或结晶的制备方法包括在碱性试剂的存在下,使n1-(2-(二甲氨基)乙基)-5-甲氧基-n1-甲基-n4-(4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)苯-1,2,4-三胺和丙烯酰氯反应的步骤。在一些具体的实施方案中,其中所述的碱性试剂优选无机碱或有机碱,所述无机碱或者有机碱选自碳酸钠、碳酸氢钠、碳酸钾、三乙胺、n,n-二异丙基乙胺中的一种或几种,进一步优选为n,n-二异丙基乙胺。在一些具体的实施方案中,本发明提供式b化合物或其水合物、溶剂合物或结晶的制备方法,其中反应溶剂为n,n-二甲基乙酰胺。所述的式c化合物或其水合物、溶剂合物或结晶的制备方法包括如下步骤:1)式c-1的化合物与甲胺反应,制得式c-2的化合物;2)式c-2的化合物与氨基保护剂反应制得式c-3的化合物,其中r1为氨基保护基;3)式c-3的化合物上的硝基经还原反应制得式c-4的化合物;4)式c-4的化合物与丙烯酰卤反应制得式c-5的化合物;5)式c-5的化合物经脱除氨基保护基生成式c的化合物。其中,所述的氨基保护剂为用于引入氨基保护基的试剂,所述的氨基保护基为如三甲基硅基、三甲基硅基乙氧基、苄氧羰基、叔丁氧羰基、2-联苯基-2-丙氧羰基、笏甲氧羰基、甲酰基、三氟乙酰基、三苯甲基、苄基等,优选为叔丁氧羰基;所述的还原反应可以在还原体系如氢气-钯/碳、铁粉/氯化铵中进行;所述的丙烯酰卤为丙烯酰氟、丙烯酰氯、丙烯酰溴、丙烯酰碘,优选为丙烯酰氯和丙烯酰溴。所述的式d化合物或其水合物、溶剂合物或结晶的制备方法包括如下步骤:1)式c-1的化合物与式d-1的化合物反应制得式d-2的化合物,其中r1为氨基保护基,具有上文所述的定义;2)式d-2的化合物上的硝基经还原反应制得式d-3的化合物;3)式d-3的化合物与式d-4的化合物反应制得式d-5的化合物,其中x1、x2各自独立地选自氟、氯、溴和碘;4)式d-5的化合物经脱除氨基保护基,然后与三氟乙酸反应生成式d的化合物。所述的式e化合物或其水合物、溶剂合物或结晶的制备方法包括使n-(2-((2-(二甲基氨基)乙基)(甲基)氨基)-4-甲氧基-5-((4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)氨基)苯基)烯丙酰胺进行氧化反应的步骤,其中所述的氧化反应通过加入氧化试剂发生,优选地,所述的氧化试剂包括但不限于双氧水和间氯过氧苯甲酸,优选为30%双氧水。所述的式f的化合物或其水合物、溶剂合物或结晶的制备方法包括使n1-(2-(二甲氨基)乙基)-5-甲氧基-n1-甲基-n4-(4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)苯-1,2,4-三胺与3-氯丙酰氯反应的步骤。所述的式g的化合物或其水合物、溶剂合物或结晶的制备方法包括如下步骤:1)式g-1的化合物与式g-2的化合物反应制得式g-3的化合物;2)式g-3的化合物与式g-4的化合物反应制得式g-5的化合物;3)式g-5的化合物与式g-6的化合物反应制得式g-7的化合物;4)式g-7的化合物经还原反应制得式g-8的化合物;5)式g-8的化合物与式g-9的化合物反应制得式g的化合物,其中x3、x4各自独立地选自氟、氯、溴和碘。在一些优选的实施例中,式g-1的化合物与式g-2的化合物在乙二醇二甲醚中,在无水氯化铝的催化下发生反应制得式g-3的化合物;式g-3的化合物与式g-4的化合物在仲丁醇中,在对甲苯磺酸一水物的催化下,发生反应制得式g-5的化合物;式g-5的化合物与式g-6的化合物在n,n-二甲基乙酰胺中,在n,n-二异丙基乙胺的作用下,发生反应制得式g-7的化合物;g-8的化合物与式g-9的化合物在n,n-二甲基乙酰胺中反应,然后在三乙胺的作用下脱除氯化氢生成反应制得式g的化合物。所述的式h的化合物或其水合物、溶剂合物或结晶的制备方法包括:在碱性试剂下,使n1-(2-(二甲氨基)乙基)-5-甲氧基-n1-甲基-n4-(4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)苯-1,2,4-三胺和乙酸酐反应的步骤。在一些具体的实施方案中,其中所述的碱性试剂优选无机碱或有机碱,所述无机碱或者有机碱选自碳酸钠、碳酸氢钠、碳酸钾、三乙胺、n,n-二异丙基乙胺中的一种或几种,进一步优选为三乙胺。所述的式i的化合物或其水合物、溶剂合物或结晶的制备方法包括如下步骤:1)式g-1的化合物与式g-2的化合物反应制得式i-1的化合物;2)式i-1的化合物与式g-4的化合物反应制得式i-2的化合物;3)式i-2的化合物与式g-6的化合物反应制得式i-3的化合物;4)式i-3的化合物经还原反应制得式i-4的化合物;5)式i-4的化合物与3-氯丙酰氯在n,n-二甲基乙酰胺中反应,然后在三乙胺的作用下脱除氯化氢,取该反应母液,使用制备色谱,分离纯化制得式i的化合物。在一些优选的实施方案中,式g-1的化合物与式g-2的化合物在无水氯化铝的催化下,在乙二醇二甲醚中,发生反应,制得式i-1的化合物;式i-1的化合物与式g-4的化合物在对甲苯磺酸一水物的催化下,在仲丁醇中,发生反应,制得式i-2的化合物;式i-2的化合物与式g-6的化合物在n,n-二异丙基乙胺的作用下,在n,n-二甲基乙酰胺中,发生反应,制得式i-3的化合物;式i-3的化合物在铁粉和氯化铵的作用下,经还原反应制得式i-4的化合物;式i-4的化合物与3-氯丙酰氯在n,n-二甲基乙酰胺中反应,然后在三乙胺的作用下脱除氯化氢,取该反应母液,使用制备色谱,分离纯化制得式i的化合物。本发明的第三个目的是提供一种定位、定量式a化合物甲烷磺酸盐中作为副产物的式b、式c、式d、式e、式f、式g、式h和式i的化合物的hplc方法。当式a化合物甲烷磺酸盐作为原料药生产时,产品中可能会存在一定量的作为副产物的式b、式c、式d、式e、式f、式g、式h和式i化合物。为满足药品的要求,需要检测和控制作为杂质的式b、式c、式d、式e、式f、式g、式h和式i化合物的含量。因此,式b、式c、式d、式e、式f、式g、式h和式i化合物可以作为参比对照品,应用于hplc法分析式a化合物甲烷磺酸盐中相关杂质的归属和定位,可以用于式a化合物甲烷磺酸盐中作为副产物的式b、式c、式d、式e、式f、式g、式h和式i化合物的定量分析以及用于式a化合物甲烷磺酸盐的质量分析。本发明的所述利用式b、式c、式d、式e、式f、式g、式h和式i化合物分析式a化合物甲烷磺酸盐的hplc方法包括以下步骤:1)制备各含有式b、式c、式d、式e、式f、式g、式h、式i、式a化合物甲烷磺酸盐的定位溶液;2)制备含有式b、式c、式d、式e、式f、式g、式h、式i和式a化合物甲烷磺酸盐的系统适用性溶液;和3)使用步骤1)制得的定位溶液和步骤2)制得的系统适用性溶液通过高效液相色谱法分离式b、式c、式d、式e、式f、式g、式h、式i和式a化合物甲烷磺酸盐,并确定式b、式c、式d、式e、式f、式g、式h、式i和式a化合物甲烷磺酸盐的位置和含量。在一些具体的实施方案中,根据本发明的利用式b、式c、式d、式e、式f、式g、式h、式i和式a化合物甲烷磺酸盐的hplc方法,其中步骤2)的所述的系统适应性溶液的制备方法包括:i)精密称定式b、式c、式d、式e、式f、式g、式h和式i化合物,分别用醇类溶剂溶解,获得八份溶液;和ii)精密称定式a化合物甲烷磺酸盐,用醇类溶剂溶解,再加入步骤i)制得的八份溶液,之后再用醇类溶剂稀释,得到系统适应性溶液,其中所述的醇类溶剂优选为甲醇、乙醇、丙醇或异丙醇,更优选为甲醇。在本文中,式b、式c、式d、式e、式f、式g、式h、式i和式a化合物甲烷磺酸盐还包括所述化合物的水合物、溶剂合物、结晶或药学上可接受的盐。附图说明图1是式a化合物甲烷磺酸盐及杂质式c、式d、式e、式g、式h化合物混合进样的hplc图谱,其中12.467min为式a化合物甲烷磺酸盐,8.286min为杂质式c化合物,7.754min为杂质式d化合物,4.898min为杂质式e化合物,12.009min为杂质式g化合物,10.029min为杂质式h化合物,13.106min为杂质f化合物。具体实施方式下面结合实施例对本发明作进一步详细阐述,但本发明不限于这些实施例。以下实施例中使用的材料如无特殊说明均为商购获得。实施例1n-(2-((2-(二甲基氨基)乙基)(甲基)氨基)-4-甲氧基-5-((4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)氨基)苯基)烯丙酰胺甲烷磺酸盐的制备步骤1:10-(2-氯嘧啶-4-基)-6,7,8,9-四氢吡啶并[1,2-a]吲哚的合成在100l立式夹套玻璃反应釜中,加入乙二醇二甲醚(39.15kg)和2,4-二氯嘧啶(3.915kg),将固液混合物降温至10℃以下,分批加入无水氯化铝(3.855kg),控制加入速度,温度不高于30℃。加入完毕,在25±5℃搅拌30分钟,然后加入6,7,8,9-四氢吡啶并[1,2-a]吲哚(4.500kg),升温,在60±5℃,反应3小时,hplc监控至6,7,8,9-四氢吡啶并[1,2-a]吲哚含量不超过1.0%,确定反应完全。将反应液降温至25℃以下,加入纯化水(90.0kg),搅拌,过滤,将滤饼加入乙腈(17.8kg)中,打浆,过滤,干燥得到黄色粉末状固体,共6.652kg,收率89.2%。步骤2:n-(4-氟-2-甲氧基-5-硝基苯基)-4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-胺的合成在500l搪玻璃反应釜中加入仲丁醇(80.82kg)、10-(2-氯嘧啶-4-基)-6,7,8,9-四氢吡啶并[1,2-a]吲哚(6.652kg)、4-氟-2-甲氧基-5-硝基苯胺(4.363kg)、对甲苯磺酸一水物(4.816kg),得到固液混合物,将反应液升温至回流,固体逐渐溶解,随着反应进行,有黄色固体析出,回流7.5小时后,hplc监控至反应完全。停止加热,将反应液降温至15℃以下,搅拌1小时,将固体离心过滤,在滤饼中加入乙腈(31.5kg),在25±5℃打浆1.5小时,离心,干燥得标题化合物,共9.548kg,收率94.0%。步骤3:n1-(2-二甲基氨基乙基)-5-甲氧基-n1-甲基-2-硝基-n4-(4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)苯基-1,4-二胺的合成在100l立式夹套玻璃反应釜中加入n,n-二甲基乙酰胺(44.7kg)、n-(4-氟-2-甲氧基-5-硝基苯基)-4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-胺(9.548kg)、n,n,n’-三甲基乙二胺(3.380kg)和n,n-二异丙基乙胺(4.841kg),氮气保护下,反应混合物在85±5℃反应2小时,hplc监控至反应完全。将反应液冷却至70℃以下,加入纯化水(95.5kg),过滤,干燥得到标题化合物,共8.206kg,收率72.2%。步骤4:n1-(2-(二甲氨基)乙基)-5-甲氧基-n1-甲基-n4-(4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)苯-1,2,4-三胺的合成在100l立式夹套反应釜中加入无水乙醇(32.39kg)、纯化水(14.32kg)、n1-(2-二甲基氨基乙基)-5-甲氧基-n1-甲基-2-硝基-n4-(4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)苯基-1,4-二胺(4.103kg)、还原铁粉(2.224kg)和氯化铵(2.129kg),反应混合物回流1.5小时后,hplc监控至反应完全。将反应液降温至50℃以下,使用硅藻土助滤,将固体滤除,浓缩滤液,向残余物中加入四氢呋喃(3.45kg)和纯化水(34.71kg),打浆,过滤,干燥得到标题化合物3.244kg,收率84.0%。步骤5:n-(2-((2-(二甲基氨基)乙基)(甲基)氨基)-4-甲氧基-5-((4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)氨基)苯基)烯丙酰胺的合成在100l立式夹套玻璃反应釜中加入n,n-二甲基乙酰胺(48.6kg),升温至40℃,将n1-(2-(二甲氨基)乙基)-5-甲氧基-n1-甲基-n4-(4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)苯-1,2,4-三胺(6.487kg)加入,然后开始滴加3-氯丙酰氯(1.777kg),控制滴加速度,不高于60℃,滴加完毕,开启降温,在40±5℃搅拌1小时后,取样hplc监控至反应完全。将纯化水(0.253kg)加入,搅拌30分钟。将反应液加热,在80±5℃的条件下,将三乙胺(13.52kg)加入,升温至95±5℃,反应2小时后,hplc检测至反应完全。降温,加入甲醇(83.0kg),然后降温析晶,过滤,干燥得标题化合物4.953kg,收率68.6%。步骤6:n-(2-((2-(二甲基氨基)乙基)(甲基)氨基)-4-甲氧基-5-((4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)氨基)苯基)烯丙酰胺甲烷磺酸盐的制备称步骤5制得的n-(2-((2-(二甲基氨基)乙基)(甲基)氨基)-4-甲氧基-5-((4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)氨基)苯基)烯丙酰胺(148.5mg,0.28mmol)溶于1.49ml95%的丙酮中,55℃时固体溶清,用1ml丙酮稀释甲磺酸(29.11mg,0.30mmol),55℃下将甲磺酸的丙酮溶液加至上述化合物(i)的丙酮溶液中,反应2h后过滤,丙酮洗涤,室温下真空干燥获得甲烷磺酸盐155mg。1hnmr(500mhz,dmso-d6):δ9.56(s,1h,重水交换后消失),9.20(s,1h,重水交换后消失),8.50(s,1h,),8.36(d,1h),8.09(d,1h),7.91(s,1h,重水交换后消失),7.43(d,1h),7.16(t,1h),7.12(t,1h),7.05(d,1h),6.97(s,1h,),6.63(dd,1h,),6.29(dd,1h),5.78(d,1h),4.11(t,2h),3.90(s,3h),3.33-3.31(m,2h),3.26-3.21(m,4h),2.81(s,6h),2.62(s,3h),2.45(s,3h),2.03-2.02(m,2h),1.84-1.83(m,2h)。esi-msm/z:540.2[m-ch3so3h+h]+。实施例2n-(5-丙烯酰胺基-4-((2-(二甲基氨基)乙基)甲基)氨基)-2-甲氧基苯基)-n-(4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)烯丙酰胺在250ml三口瓶中加入n1-(2-(二甲氨基)乙基)-5-甲氧基-n1-甲基-n4-(4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)苯-1,2,4-三胺(1.60g,3.3mmol,1.0eq),n,n-二甲基乙酰胺(16ml)、丙烯酰氯(0.90g,9.9mmol,3.0eq)、n,n-二异丙基乙胺(2.13g,16.5mmol,5.0eq),混合物在室温搅拌反应1.5小时。向反应混合物中加入50ml甲醇,析出固体,过滤,真空干燥得到标题化合物,共0.60g,收率30.0%。1hnmr(300mhz,dmso-d6)δ10.03(s,1h),8.59(d,j=5.5hz,1h),8.19(s,1h),7.91(d,j=8.0hz,1h),7.45-7.42(m,2h),7.17(d,j=7.0hz,1h),7.09-7.06(m,2h),6.77(dd,j1=17.0hz,j2=10.3hz,1h),6.42(dd,j1=17.0hz,j2=10.0hz,1h),6.29(d,j=16.0hz,1h),6.20(d,j=16.5hz,1h),5.75-5.73(m,2h),4.09(t,j=5.5hz,2h),3.71(s,3h),3.01-2.99(m,2h),2.93-2.92(m,2h),2.79(s,3h),2.43(t,j=5.5hz,2h),2.24(s,6h),2.00-1.98(m,2h),1.80(m,2h).esi-msm/z:594.3[m+h]+。实施例3n-(4-甲氧基-2-(甲基氨基)-5-((4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)氨基)苯基)烯丙酰胺步骤1:2-甲氧基-n4-甲基-5-硝基-n1-(4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)苯基-1,4-二胺的制备将n-(4-氟-2-甲氧基-5-硝基苯基)-4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-胺(6.00g,13.8mmol,1.0eq)、甲胺四氢呋喃溶液(50ml,100mmol,7.2eq)和n,n-二甲基乙酰胺(60ml)加入到反应瓶中,搅拌下室温反应过夜。次日后处理得5.50g标题化合物。步骤2:(5-甲氧基-2-硝基-4-((4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)氨基)苯基)(甲基)氨基甲酸叔丁酯的制备取步骤1制得的2-甲氧基-n4-甲基-5-硝基-n1-(4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)苯基-1,4-二胺(2.00g,4.5mmol,1.0eq)、boc酸酐(2.95g,13.5mmol,3.0eq)、n,n-二异丙基乙胺(1.45g,11.3mmol,2.5eq)及1,4-二氧六环(50ml)加入到反应瓶中,混合物在100℃反应5小时。冷却至室温,后处理得2.65g固体。步骤3:(5-甲氧基-2-氨基-4-((4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)氨基)苯基)(甲基)氨基甲酸叔丁酯的制备将步骤2制得的(5-甲氧基-2-硝基-4-((4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)氨基)苯基)(甲基)氨基甲酸叔丁酯(2.30g,4.2mmol)、钯碳(0.23g)、甲醇(5ml)及二氯甲烷(5ml)加入50ml三口烧瓶,室温下常压氢化46小时,将钯碳滤出,滤液经后处理得到棕色油状标题化合物2.30g。步骤4:(5-甲氧基-2-丙烯酰氨基-4-((4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)氨基)苯基)(甲基)氨基甲酸叔丁酯的制备将步骤3制得的(5-甲氧基-2-氨基-4-((4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)氨基)苯基)(甲基)氨基甲酸叔丁酯(2.30g,3.6mmol,1.0eq)、n,n-二异丙基乙胺(0.68g,5.4mmol,1.5eq)加入到25ml三口烧瓶中,将丙烯酰氯(0.32g,3.6mmol,1.0eq)溶解在二氯甲烷(5ml)中,滴加到冰水浴冷却的上述反应混合物中,滴加完毕,在冰水浴中继续搅拌2小时,经后处理得2.80g固体。步骤5:n-(4-甲氧基-2-(甲基氨基)-5-((4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)氨基)苯基)烯丙酰胺的制备将步骤4制得的(5-甲氧基-2-丙烯酰氨基-4-((4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)氨基)苯基)(甲基)氨基甲酸叔丁酯(2.80g)、三氟乙酸(20ml)和二氯甲烷(60ml)加入到250ml三口烧瓶中,在冰浴冷却下搅拌0.5小时,经后处理纯化得标题化合物共0.50g。1hnmr(300mhz,dmso-d6)δ9.30(s,1h),8.27(d,j=5.5hz,1h),8.07(d,j=8.0hz,1h),7.80(s,1h),7.71(s,1h),7.41(d,j=8.0hz,1h),7.16-7.09(m,2h),6.94(d,j=5.5hz,1h),6.45(dd,j1=17.0hz,j2=10.5hz,1h),6.34(s,1h),6.19(dd,j1=17.0hz,j2=2.0hz,1h),5.68(dd,j1=10.0hz,j2=2.0hz,1h),4.89(s,1h),4.09(t,j=6.0hz,2h),3.84(s,3h),3.18(t,j=6.0hz,2h),2.79(s,3h),2.02-1.99(m,2h),1.83-1.81(m,2h).esi-msm/z:469.2[m+h]+.实施例4n-(4-甲氧基-2-(甲基(甲氨基甲基)氨基)-5-((4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)氨基))烯丙酰胺三氟乙酸盐步骤1:(((5-甲氧基-2-硝基-4-((4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)氨基)苯基)(甲基)氨基)甲基)(甲基)氨基甲酸叔丁酯的制备将n-(4-氟-2-甲氧基-5-硝基苯基)-4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-胺(7.20g,16.6mmol,1.0eq)、boc保护的二甲基乙二胺(4.69g,24.9mmol,1.5eq)、n,n-二异丙基乙胺(3.65g,28.2mmol,1.7eq)及n,n-二甲基乙酰胺(35ml),升温至95℃,反应3小时。后处理得红色油状标题化合物。步骤2:n-(4-甲氧基-2-(甲基(甲氨基甲基)氨基)-5-((4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)氨基))烯丙酰胺三氟乙酸盐的制备制备方法同实施例3步骤3-5的制备方法,不同的是将原料(5-甲氧基-2-硝基-4-((4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)氨基)苯基)(甲基)氨基甲酸叔丁酯替换为(((5-甲氧基-2-硝基-4-((4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)氨基)苯基)(甲基)氨基)甲基)(甲基)氨基甲酸叔丁酯,制得标题化合物,纯度为96.23%。1hnmr(300mhz,dmso-d6)δ9.39(s,1h),9.33(s,1h),8.72(s,1h),8.71(s,1h),8.37(s,1h),8.29(d,j=6.5hz,1h),8.09(d,j=7.5hz,1h),7.48(d,j=8.0hz,1h),7.23-7.19(m,2h),7.15(dd,j1=7.5hz,j2=7.0hz,1h),7.02(s,1h),6.69(dd,j1=17.0hz,j2=10.5hz,1h),6.25(dd,j1=17.0hz,j2=2.0hz,1h),5.75(dd,j1=10.5hz,j2=1.0hz,1h),4.13(t,j=6.0hz,2h),3.84(s,3h),3.28(t,j=6.0hz,2h),3.20(t,j=6.0hz,2h),3.15(t,j=5.5hz,2h),2.63(s,6h),2.03-2.01(m,2h),1.84-1.82(m,2h).esi-msm/z:526.3[m-cf3cooh+h]+.实施例52-((2-烯丙酰胺基-5-甲氧基-4-((4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)氨基)苯基)(甲基)氨基)-n,n-二甲基乙烷-1-氨氧化物将n-(2-((2-(二甲基氨基)乙基)(甲基)氨基)-4-甲氧基-5-((4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)氨基)苯基)烯丙酰胺(3.00g)及30%双氧水加入到100ml三口烧瓶,室温搅拌1.5小时,经后处理、纯化后得到标题化合物,共0.58g,纯度为98.37%。1hnmr(300mhz,dmso-d6)δ11.93(s,1h),8.67(s,1h),8.32(d,j=5.0hz,1h),8.07(d,j=7.5hz,1h),7.89(s,1h),7.41(d,j=7.5hz,1h),7.16-7.10(m,2h),7.06(m,1h),7.01(d,j=5.5hz,1h),6.93(s,1h),6.12(dd,j1=18.0hz,j2=2.0hz,1h),5.58(dd,j1=11.3hz,j2=1.5hz,1h),4.09(t,j=6.0hz,2h),3.83(s,3h),3.44-3.43(m,2h),3.27-3.24(m,8h),3.20(t,j=6.0hz,2h),2.71(s,3h),2.00(m,2h),1.82-1.81(m,2h).esi-msm/z:556.2[m+h]+.实施例63-氯-n-(2-((2-二甲氨基乙基)(甲基)氨基)-4-甲氧基-5-((4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)氨基)苯基)丙酰胺将n1-(2-(二甲氨基)乙基)-5-甲氧基-n1-甲基-n4-(4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)苯-1,2,4-三胺(0.70g,1.44mmol,1.0eq)、n,n-二甲基乙酰胺(7ml)及3-氯丙酰氯(0.18g,1.44mmol.1.0eq)加入到50ml三口烧瓶中,室温反应1.5小时。将析出的固体滤出,真空干燥得标题化合物共0.30g,纯度为95.16%。1hnmr(300mhz,dmso-d6)δ10.90(s,1h),9.96(s,1h),8.26(d,j=6.0hz,1h),8.14-8.11(m,2h),7.49(d,j=7.5hz,1h),7.25-7.20(m,3h),6.99(s,1h),4.12(t,j=5.5hz,2h),3.87-3.83(m,5h),3.39-3.35(m,4h),3.15-3.13(m,4h),2.75(s,3h),2.74(s,3h),2.68(s,3h),2.00(m,2h),1.83(m,2h).esi-msm/z:576.3[m+h]+.实施例7n-(2-((2-二甲氨基乙基)(甲基)氨基)-4-甲氧基-5-((2-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-4-基)氨基)苯基)烯丙酰胺步骤1:10-(4-氯嘧啶-2-基)-6,7,8,9-四氢吡啶并[1,2-a]吲哚的合成将2,4-二氯嘧啶(43.52g,292mmol,1.0eq)、三氯化铝(42.82g,321mmol,1.1eq)、乙二醇二甲醚(500ml)加入2l三口瓶中,在35℃以下,搅拌0.5小时。加入6,7,8,9-四氢吡啶并[1,2-a]吲哚(50.00g,292mmol,1.0eq),将反应混合物升温至60℃搅拌3小时。然后将反应液缓慢加入到1000ml水中,控制加速速度,使温度不得过35℃,将析出固体的固体滤出,使用硅胶柱层析纯化得标题化合物共1.0g。步骤2:n-(2-((2-二甲氨基乙基)(甲基)氨基)-4-甲氧基-5-((2-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-4-基)氨基)苯基)烯丙酰胺的制备制备方法同实施例1步骤2-5的制备方法,不同的是将原料10-(2-氯嘧啶-4-基)-6,7,8,9-四氢吡啶并[1,2-a]吲哚替换成步骤1中制得的10-(4-氯嘧啶-2-基)-6,7,8,9-四氢吡啶并[1,2-a]吲哚,制得标题化合物,纯度为75.59%。1hnmr(300mhz,dmso-d6)δ10.06(s,1h),8.58(s,1h),8.51(s,1h),8.41(d,j=8.0hz,1h),8.27(d,j=5.5hz,1h),7.34(d,j=8.0hz,1h),7.10(t,j=7.5hz,1h),7.04(t,j=7.5hz,1h),7.00(s,1h),6.54-6.48(m,2h),6.21(d,j=17.0hz,1h),5.75(d,j=11.0hz,1h),4.04(t,j=6.0hz,2h),3.81(s,3h),3.27(t,j=6.0hz,2h),3.00(t,2h),2.69(s,3h),2.54(s,2h),2.32(s,6h),1.96-1.95(m,2h),1.76-1.75(m,2h).esi-msm/z:540.3[m+h]+.实施例8n-(2-((2-二甲氨基乙基)(甲基)氨基)-4-甲氧基-5-((4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)氨基)苯基)乙酰胺将n1-(2-(二甲氨基)乙基)-5-甲氧基-n1-甲基-n4-(4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)苯-1,2,4-三胺(2.00g,4.12mmol,1.0eq)、乙酸酐(0.50g,4.94mmol,1.2eq)、三乙胺(0.83g,8.24mmol,2.0eq)及二氯甲烷20ml加入100ml三口瓶中,控温25℃,搅拌约2.0h。停止反应,经后处理得到1.80g棕色固体。取1.50g固体加入15ml二氯甲烷溶解,搅拌下加入30ml庚烷,0℃析晶1h,过滤得类白色滤饼,干燥得0.95g固体,收率63.3%,纯度为99.00%。1hnmr(300mhz,dmso-d6)δ9.92(s,1h),8.61(s,1h),8.32(d,j=5.0hz,1h),8.08(d,j=7.5hz,1h),7.97(s,1h),7.42(d,j=7.0hz,1h),7.16(dd,j1=j2=7.5hz,1h),7.11(dd,j1=j2=7.5hz,1h),7.00(d,j=5.0hz,1h),6.97(s,1h),4.10(s,2h),3.80(s,3h),3.19(s,2h),2.86(s,2h),2.70(s,3h),2.30(s,2h),2.22(s,6h),2.01(s,5h),1.82(s,2h).esi-msm/z:528.3[m+h]+.实施例9n-(2-((2-二甲基氨基乙基)(甲基)氨基)-4-甲氧基-5-((4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)氨基)苯基)-n-(3-((2-((2-二甲氨基乙基)(甲基)氨基)-4-甲氧基-5-((4-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基)嘧啶-2-基)氨基)苯基)氨基)-3-氧代丙基)烯丙酰胺取实施例1步骤5的反应母液,使用制备色谱,分离纯化得标题化合物0.45g,纯度87.88%,1hnmr(300mhz,dmso-d6)δ10.02(bs,1h),9.26(bs,1h),8.42(s,1h),8.31-8.28(m,2h),8.08-8.07(m,2h),7.94(s,1h),7.71(d,j=8.0hz,1h),7.46(d,j=8.0hz,1h),7.29-7.25(m,1h),7.22(t,j=7.5hz,1h),7.14-7.10(m,2h),7.06-7.03(t,j=7.5hz,2h),6.98-6.97(d,j=5.5hz,1h),6.94(s,2h),6.80-6.75(m,1h),6.25(dd,j1=17hz,j2=1.5hz,1h),5.77(d,j=11.5hz,1h),4.60(s,2h),4.06(t,j=6.0hz,2h),3.96(s,2h),3.86(s,3h),3.74(s,3h),3.35-3.20(m,10h),3.16-3.14(t,j=6.0hz,2h),2.90(s,2h),2.83-2.82(m,12h),2.72(s,3h),2.61(s,3h),1.98-1.97(m,2h),1.94-1.93(m,2h),1.80(m,2h),1.65(m,2h).esi-msm/z:1079.6[m+h]+.实验例1:式c、式d、式e、式f、式g、式h化合物的定位分析混合溶液的配制:精密称取杂质式c、式d、式e、式f、式g、式h化合物及式a化合物甲烷磺酸盐对照品适量,用甲醇溶解,配制成每1ml含500μg式a化合物甲烷磺酸盐,含各杂质1μg的溶液。将混合溶液进样分析,使用的色谱柱为agilenteclipseplusc18柱(4.6*150mm,5μm),流动相为水相:10mmol/l的磷酸二氢钾溶液(氢氧化钾调ph至7.5)、有机相:乙腈,流速为1.0ml/min;柱温为40℃,洗脱梯度缩短时间如下表1:表1时间(min)水相(%)有机相(%)0604015158515.16040256040使用紫外检测器在260nm波长处确定位置和含量。实验结果显示,在图1中12.467min为式a化合物甲烷磺酸盐,8.286min为杂质式c化合物,7.754min为杂质式d化合物,4.898min为杂质式e化合物,12.009min为杂质式g化合物,10.029min为杂质式h化合物,13.106min为杂质f化合物,具体见图1。尽管以上已经对本发明作了详细描述,但是本领域技术人员理解,在不偏离本发明的精神和范围的前提下可以对本发明进行各种修改和改变。本发明的权利范围并不限于上文所作的详细描述,而应归属于权利要求书。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1