一种钯催化8-甲基喹啉衍生物与α-酮酸的脱氢偶联反应合成α-酮酸酯的方法与流程
本发明涉及一种α-酮酸酯衍生物的合成方法,特别涉及一种钯催化α-甲基喹啉衍生物与α-酮酸的脱氢偶联反应合成α-酮酸酯衍生物的方法,属于医药中间体合成、精细有机合成领域。
背景技术:
α-酮酸酯衍生物是重要的有机化合物之一,广泛的应用于药物(如下分子结构式)、天然产物等的合成。然而合成α-酮酸酯衍生物的方法还极为有限,主要的传统方法是通过friedel-crafts酰化反应,氧化反应或水解反应合成。这些方法通常需要大量对环境不利的试剂和苛刻的反应条件,且底物适用范围有限[(a)r.deshidi,s.devari,b.a.shah,eur.j.org.chem.2015,2015,1428;(b)s.r.reddy,s.stella,a.chadha,synth.commun.2012,42,3493;(c)s.k.alamsetti,g.sekar,chem.commun.2010,46,7235;(d)j.t.moon,j.y.jeon,h.a.park,y.s.noh,k.t.lee,j.kim,d.j.choo,j.y.lee,bioorg.med.chem.lett.2010,20,734;(e)j.m.xiang,b.l.li,chin.chem.lett.2009,20,55;(f)j.lou,c.gao,y.ma,l.huang,l.li,tetrahedronlett.2006,47,311;(g)t.nopporn,p.vilailak,t.sopchok,r.somsak,tetrahedronlett.2003,44,1019;(h)p.a.manís,m.w.j.rathke,org.chem.1980,45,4952;(i)e.j.corey,b.w.erickson,j.org.chem.1971,36,3553.]。最近,kim开发了钌催化的二氢吲哚c(sp2)-h键与乙二醛酸乙酯的脱氢偶联。该反应以cu(oac)2作为氧化剂和agsbf6作为添加剂进行,并且实现了令人满意的区域选择性[h.jo,j.park,n.k.mishra,m.jeon,s.sharma,h.oh,s.y.lee,y.h.jung,i.s.kim,tetrahedron2017,73,1725]。wang及其同事报道了铜催化的α-酮酸与苯乙酮或苯丙酮的直接脱氢偶合。guo还报道了铜催化直接α-酮酸酯化的例子。然而,使用铜催化剂的这两种方法仅适用于芳香族酮,并且需要120℃的高反应温度[(a)j.du,x.zhang,x.sun,l.wang,chem.commun.2015,51,4372;(b)j.w.xu,p.y.ji,y.f.liu,w.p.luo,q.liu,c.c.guo,eur.j.org.chem.2017,2017,734.]。zhang和同事报道了fe催化的苄基c-h键与α-酮酸的直接脱氢偶合。在该策略中,使用潜在爆炸性的二叔丁基过氧化物(dtbp)作为氧化剂,并且需要100℃的高反应温度。只有甲苯衍生物适用于该反应[y.he,j.mao,g.rong,h.yan,g.zhang,adv.synth.catal.2015,357,2125.]。因此,开发一种操作简单、条件温和的合成α-酮酸酯衍生物的新方法具有重要的理论意义和应用价值。
技术实现要素:
针对现有的合成α-酮酸酯衍生物的方法存在的不足,如需要大量对环境不利的试剂,苛刻的反应条件,潜在爆炸性的过氧化物作为氧化剂等问题,本发明的目的是在于提供一种钯催化8-甲基喹啉衍生物与α-酮酸的脱氢偶联反应合成α-酮酸酯的方法,该方法具有反应条件温和、一步反应、与空气相容、可扩量反应等优点。目前,基于8-甲基喹啉衍生物与α-酮酸的脱氢偶联反应合成α-酮酸酯的方法尚未见报道。因此,该方法在α-酮酸酯衍生物的合成应用领域具有很好的应用前景。
为了实现上述技术目的,本发明提供了一种钯催化8-甲基喹啉衍生物与α-酮酸的脱氢偶联反应合成α-酮酸酯的方法,该方法是:在70℃,空气条件下,式1取代喹啉和式2取代α-酮酸一锅反应,得到式3α-酮酸酯衍生物;
其中,
ar选自苯、噻吩、或萘;
r1、r2和r3独立选自氢或其他取代基。
优选的方案,式1取代8-甲基喹啉为8-甲基喹啉、7,8-二甲基喹啉、6,8-二甲基喹啉、7-甲氧基-8-甲基喹啉、5-氟-8-甲基喹啉、7-氟-8-甲基喹啉、7-氯-8-甲基喹啉、6-溴-8-甲基喹啉、8-甲基-5-硝基喹啉、8-甲基-6-硝基喹啉、8-乙基喹啉、8-苄基喹啉。
优选的方案,式2取代α-酮酸为苯甲酰甲酸、2-氧代-2-(邻甲苯基)乙酸、2-氧代-2-(对甲苯基)乙酸、2-(4-甲氧基苯基)-2-氧代乙酸、2-(2-氟苯基)-2-氧代乙酸、2-(4-氯苯基)-2-氧代乙酸、2-(3-氯苯基)-2-氧代乙酸、2-(2-溴苯基)-2-氧代乙酸、2-(4-溴苯基)-2-氧代乙酸、2-(萘-1-基)-2-氧代乙酸、2-氧代-2-(噻吩-2-基)乙酸。
优选的方案,所述8-甲基喹啉衍生物与α-酮酸通过钯催化脱氢偶联反应转化成式3α-酮酸酯衍生物,这些α-酮酸酯衍生物为喹啉-8-甲基-2-氧代-2-苯基乙酸酯、(7-甲基喹啉-8-基)甲基-2-氧代-2-苯基乙酸酯、(6-甲基喹啉-8-基)甲基-2-氧代-2-苯基乙酸酯、(7-甲氧基喹啉-8-基)甲基-2-氧代-2-苯基乙酸酯、(5-氟喹啉-8-基)甲基-2-氧代-2-苯基乙酸酯、(7-氟喹啉-8-基)甲基-2-氧代-2-苯基乙酸酯、(7-氯喹啉-8-基)甲基-2-氧代-2-苯基乙酸酯、(6-溴喹啉-8-基)甲基-2-氧代-2-苯基乙酸酯、(5-硝基喹啉-8-基)甲基-2-氧代-2-苯基乙酸酯、(6-硝基喹啉-8-基)甲基-2-氧代-2-苯基乙酸酯、1-(喹啉-8-基)乙基-2-氧代-2-苯基乙酸酯、苯基(喹啉-8-基)甲基-2-氧代-2-苯基乙酸酯、喹啉-8-基甲基-2-氧代-2-(邻-甲苯基)乙酸酯、喹啉-8-基甲基-2-氧代-2-(对甲苯基)乙酸酯、喹啉-8-基甲基-2-氧代-2-(4-甲氧苯基)乙酸酯、喹啉-8-基甲基-2-氧代-2-(2-氟苯基)乙酸酯、喹啉-8-基甲基-2-氧代-2-(4-氯苯基)乙酸酯、喹啉-8-基甲基-2-氧代-2-(3-氯苯基)乙酸酯、喹啉-8-基甲基-2-氧代-2-(2-溴苯基)乙酸酯、喹啉-8-基甲基-2-氧代-2-(4-溴苯基)乙酸酯、喹啉-8-基甲基-2-氧代-2-(噻吩-2-基)乙酸酯喹啉-8-基甲基-2-氧代-2-(萘-1-基)乙酸酯。
优选的方案,钯催化剂为醋酸钯、氯化钯、二苯腈合二氯化钯、三(二亚苄基丙酮)二钯、二(氰基苯)二氯化钯、双三苯基磷二氯化钯、四(三苯基膦)钯、(1,5-环辛二烯)二氯化钯中至少一种;较优选为三(二亚苄基丙酮)二钯、醋酸钯或二苯腈合二氯化钯;最优选为三(二亚苄基丙酮)二钯。
优选的方案,氧化剂为过硫酸钾、高碘酸钠、过硫酸铵、醋酸铜、醋酸碘苯或叔丁基过氧化氢中至少一种;较优选为醋酸碘苯或过硫酸钾;最优选的氧化剂为醋酸碘苯。
优选的方案,三(二亚苄基丙酮)二钯为催化剂、其用量为0.05~0.2当量、时间为10~18小时、反应温度60~80℃。进一步优选的方案、醋酸碘苯用量为1.5~3当量、时间为10~18小时、反应温度60~80℃。三(二亚苄基丙酮)二钯和氧化剂用量过低、产率下降。反应时间过短产率降低、而反应时间过长产率无明显变化。在优选的反应时间和温度范围内可以达到最佳的反应效果。
优选的方案,反应中采用的溶剂为三氯甲烷、水、n,n-二甲基甲酰胺、苯、二氯甲烷、二氧六环、丙酮、硝基甲烷、乙酸乙酯中至少一种,较优选为甲苯、二氯甲烷、三氯甲烷;最优选为三氯甲烷。
本发明的α-酮酸酯衍生物合成中反应方程式如下。
基于大量的实验总结以及参考先前文献报道,本发明提出了如下合理的反应机制。首先,8-甲基喹啉衍生物的苄基c-h键被钯催化剂活化产生中间体a;中间体a被phi(o2ccoar)2氧化成pd(ii)物种b,这是由phi(oac)2和α-酮酸之间的配体交换反应产生的。最后,中间体b通过还原消除实现所需的α-酮酸酯,并再生pd(0)物质。
本发明的技术方案中,8-甲基喹啉衍生物0.1毫摩尔、α-酮酸衍生物0.2毫摩尔、二钯三(二亚苄基丙酮)二钯0.01毫摩尔、二乙酸碘苯0.25毫摩尔将其溶于1毫升三氯甲烷,在空气氛围下放入设定温度的热浴锅磁力搅拌反应12小时。反应冷却至室温,加入饱和碳酸氢钠溶液中和。然后用乙酸乙酯萃取;合并萃取液并用无水硫酸钠干燥,过滤,滤液经减压浓缩后,以石油醚:乙酸乙酯=4:1为洗脱剂对粗产物进行柱层析得到纯品。
本发明的α-酮酸酯衍生物的合成方法,包括以下步骤:
称取8-甲基喹啉衍生物0.1毫摩尔、α-酮酸衍生物0.2毫摩尔、二钯三(二亚苄基丙酮)二钯0.01毫摩尔、二乙酸碘苯0.25毫摩尔将其溶于1毫升三氯甲烷,在空气氛围下放入设定温度的热浴锅磁力搅拌反应12小时。反应冷却至室温,加入饱和碳酸氢钠溶液中和。然后用乙酸乙酯萃取;合并萃取液并用无水硫酸钠干燥,过滤,滤液经减压浓缩后,以石油醚:乙酸乙酯=4:1为洗脱剂对粗产物进行柱层析得到纯品。
相对现有技术,本发明的技术方案具有以下优点和效果:
1)本发明的技术方案首次以三(二亚苄基丙酮)二钯为催化剂、经由8-甲基喹啉衍生物与α-酮酸的脱氢偶联反应一步实现了α-酮酸酯衍生物的制备。
2)本发明的技术方案反应条件温和,操作简单。
3)本发明的技术方案避免使用大量对环境不利的试剂和潜在爆炸性的过氧化物。
4)本发明的技术方案在温和条件下实现了α-酮酸酯衍生物的一步合成,反应对空气及水兼容,在低钯催化剂负载量(1mol%)下容易地扩增至克水平,具有步骤简单、成本低、操作简单等优点、克服了现有技术如反应试剂毒性大、反应步骤多、副产物多等缺陷。
附图说明
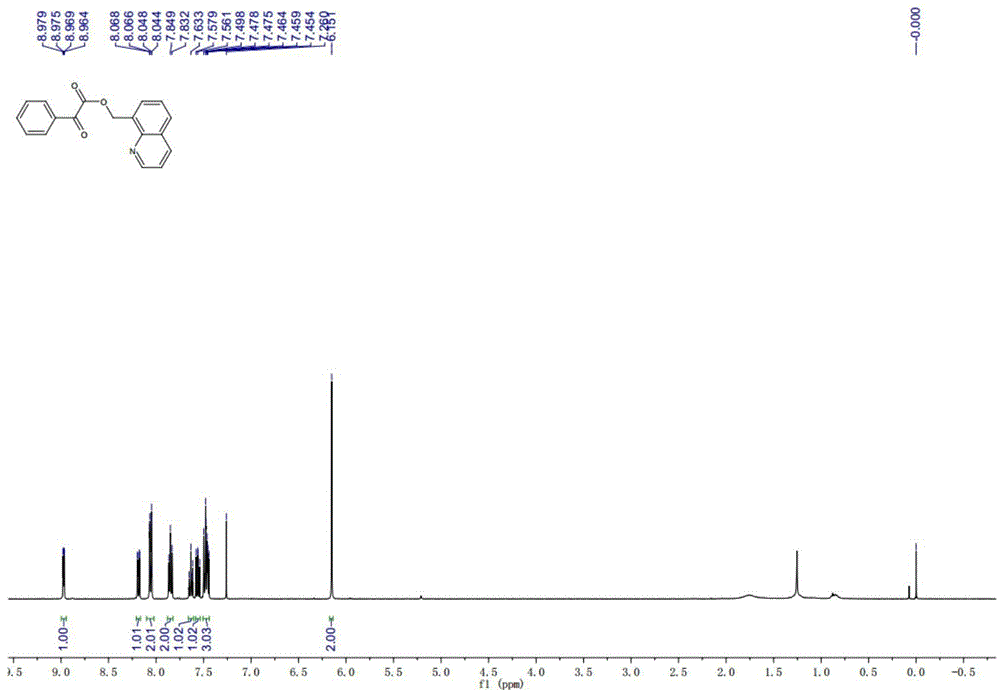
【图1】为实施例1所得产物的1hnmr图;
【图2】为实施例1所得产物的13cnmr图;
【图3】为实施例13所得产物的1hnmr图;
【图4】为实施例13所得产物的13cnmr图。
具体实施方式
结合以下具体实施例和附图,对本发明作进一步的详细说明。实施本发明的过程、条件、试剂、试验方法等、除以下专门提及的内容之外,均为本领域的普遍和公知常识,本发明没有特别限制内容。
实施例1
将8-甲基喹啉(14.3mg,0.1mmol)、苯甲酰甲酸(30.0mg,0.2mmol)、三(二亚苄基丙酮)二钯(9.2mg,0.01mmol)、二乙酸碘苯(80.5mg,0.25mmol)、及一粒搅拌子放入反应管中,加入1毫升溶剂chcl3,密封反应管。将反应管置于70℃油浴反应锅中搅拌反应12小时。冷至室温后,用饱和碳酸氢钠溶液中和并用乙酸乙酯萃取。合并萃取液并用无水硫酸钠干燥、过滤、滤液经减压浓缩后,以乙酸乙酯:石油醚=1:4(体积比)为洗脱剂对粗产物进行柱层析得到纯品。白色固体,熔点71-72℃,产率80%。1hnmr(400mhz,cdcl3)δ8.97(dd,j=4.2,1.8hz,1h),8.18(dd,j=8.2,1.8hz,1h),8.07-8.04(m,2h),7.85(t,j=6.6hz,2h),7.65-7.62(m,1h),7.56(dd,j=8.2,7.0hz,1h),7.50-7.44(m,3h),6.15(s,2h);13cnmr(100mhz,cdcl3)δ186.48,163.92,150.05,146.19,136.27,134.88,132.91,132.59,130.22,129.26,128.86,128.77,128.26,126.27,121.52,64.44;ir(kbr)3023,2947,1727,1686,1680,1449,1402,1318,1176,993,828,790cm-1;hrms(esi):m/z[m+h]+calcdforc18h14no3:292.0968,found:292.0969.
实施例2
将7,8-二甲基喹啉(15.7mg,0.1mmol)、苯甲酰甲酸(30.0mg,0.2mmol)、三(二亚苄基丙酮)二钯(9.2mg,0.01mmol)、二乙酸碘苯(80.5mg,0.25mmol)、及一粒搅拌子放入反应管中,加入1毫升溶剂chcl3,密封反应管。将反应管置于70℃油浴反应锅中搅拌反应12小时。冷至室温后,用饱和碳酸氢钠溶液中和并用乙酸乙酯萃取。合并萃取液并用无水硫酸钠干燥、过滤、滤液经减压浓缩后,以乙酸乙酯:石油醚=1:4(体积比)为洗脱剂对粗产物进行柱层析得到纯品。白色固体,熔点118-119℃,产率71%。1hnmr(400mhz,cdcl3)δ8.96(s,1h),8.12(d,j=8.0hz,1h),8.03(d,j=7.6hz,2h),7.76(d,j=8.4hz,1h),7.60(t,j=7.2hz,1h),7.46-7.37(m,4h),6.25(s,2h),2.67(s,3h);13cnmr(100mhz,cdcl3)δ186.67,164.30,150.15,147.22,140.50,136.03,134.76,132.58,130.16,129.75,129.56,128.85,128.76,126.62,120.63,60.73,19.97;ir(kbr)3465,3174,2359,1731,1681,1402,1195,1176,980,943,688cm-1;hrms(esi):m/z[m+nh4]+calcdforc19h19n2o3:323.1390,found:323.1387.
实施例3
将6,8-二甲基喹啉(15.7mg,0.1mmol)、苯甲酰甲酸(30.0mg,0.2mmol)、三(二亚苄基丙酮)二钯(9.2mg,0.01mmol)、二乙酸碘苯(80.5mg,0.25mmol)、及一粒搅拌子放入反应管中,加入1毫升溶剂chcl3,密封反应管。将反应管置于70℃油浴反应锅中搅拌反应12小时。冷至室温后,用饱和碳酸氢钠溶液中和并用乙酸乙酯萃取。合并萃取液并用无水硫酸钠干燥、过滤、滤液经减压浓缩后,以乙酸乙酯:石油醚=1:4(体积比)为洗脱剂对粗产物进行柱层析得到纯品。黄色油状,产率75%。1hnmr(400mhz,cdcl3)δ8.90(dd,j=4.0,1.6hz,1h),8.10-8.05(m,3h),7.68-7.60(m,3h),7.48(t,j=7.8hz,2h),7.41(dd,j=8.2,4.2hz,1h),6.11(s,2h),2.54(s,3h);13cnmr(100mhz,cdcl3)δ186.54,163.94,149.17,144.78,136.14,135.59,134.92,132.43,131.66,130.24,128.86,128.38,127.55,121.53,64.48,21.66;ir(kbr)3140,2363,1737,1597,1498,1400,1197,1176,980,866,710,686cm-1;hrms(esi):m/z[m+na]+calcdforc19h15nnao3:328.0944,found:328.0942.
实施例4
将7-甲氧基-8-甲基喹啉(17.3mg,0.1mmol)、苯甲酰甲酸(30.0mg,0.2mmol)、三(二亚苄基丙酮)二钯(9.2mg,0.01mmol)、二乙酸碘苯(80.5mg,0.25mmol)、及一粒搅拌子放入反应管中,加入1毫升溶剂chcl3,密封反应管。将反应管置于70℃油浴反应锅中搅拌反应12小时。冷至室温后,用饱和碳酸氢钠溶液中和并用乙酸乙酯萃取。合并萃取液并用无水硫酸钠干燥、过滤、滤液经减压浓缩后,以乙酸乙酯:石油醚=1:4(体积比)为洗脱剂对粗产物进行柱层析得到纯品。黄色油状,产率77%。1hnmr(400mhz,cdcl3)δ8.94(dd,j=4.2,1.8hz,1h),8.11-8.05(m,3h),7.87(d,j=9.2hz,1h),7.61-7.58(m,1h),7.45(t,j=7.8hz,2h),7.37(d,j=9.2hz,1h),7.30(dd,j=8.2,4.2hz,1h),6.20(s,2h),4.04(s,3h);13cnmr(100mhz,cdcl3)δ186.97,164.43,159.16,151.03,147.79,136.07,134.63,132.73,130.66,130.22,128.69,123.27,119.28,117.18,113.48,58.24,56.52;ir(kbr)3438,1832,1716,1683,1649,1457,1402,1320,1141,1097,684cm-1;hrms(esi):m/z[m+nh4]+calcdforc19h19n2o4:339.1339,found:339.1329.
实施例5
将5-氟-8-甲基喹啉(16.1mg,0.1mmol)、苯甲酰甲酸(30.0mg,0.2mmol)、三(二亚苄基丙酮)二钯(9.2mg,0.01mmol)、二乙酸碘苯(80.5mg,0.25mmol)、及一粒搅拌子放入反应管中,加入1毫升溶剂chcl3,密封反应管。将反应管置于70℃油浴反应锅中搅拌反应12小时。冷至室温后,用饱和碳酸氢钠溶液中和并用乙酸乙酯萃取。合并萃取液并用无水硫酸钠干燥、过滤、滤液经减压浓缩后,以乙酸乙酯:石油醚=1:4(体积比)为洗脱剂对粗产物进行柱层析得到纯品。黄色固体,熔点78-79℃,产率80%。1hnmr(400mhz,cdcl3)δ8.94(s,1h),8.38(d,j=8.4hz,1h),7.97(d,j=7.2hz,2h),7.74(t,j=6.4hz,1h),7.56(t,j=7.2hz,1h),7.46-7.39(m,3h),7.16(t,j=8.6hz,1h),6.00(s,2h);13cnmr(100mhz,cdcl3)δ186.37,163.86,159.48,156.93,150.91,146.76,134.93,132.50,130.18,129.59,129.54,129.49,128.86,121.60,121.58,119.13(d,j=16.5hz),109.77(d,j=19.4hz),63.99;19fnmr(377mhz,cdcl3)δ-121.06;ir(kbr)3138,1735,1686,1597,1579,1402,1321,1194,1176,988,833,811,688cm-1;hrms(esi):m/z[m+h]+calcdforc18h13fno3:310.0874,found:310.0876.
实施例6
将7-氟-8-甲基喹啉(16.1mg,0.1mmol)、苯甲酰甲酸(30.0mg,0.2mmol)、三(二亚苄基丙酮)二钯(9.2mg,0.01mmol)、二乙酸碘苯(80.5mg,0.25mmol)、及一粒搅拌子放入反应管中,加入1毫升溶剂chcl3,密封反应管。将反应管置于70℃油浴反应锅中搅拌反应12小时。冷至室温后,用饱和碳酸氢钠溶液中和并用乙酸乙酯萃取。合并萃取液并用无水硫酸钠干燥、过滤、滤液经减压浓缩后,以乙酸乙酯:石油醚=1:4(体积比)为洗脱剂对粗产物进行柱层析得到纯品。白色固体,熔点141-143℃,产率68%。1hnmr(400mhz,cdcl3)δ9.00(dd,j=4.0,1.6hz,1h),8.17(dd,j=8.2,1.8hz,1h),8.06-8.04(m,2h),7.88(dd,j=8.8,6.0hz,1h),7.62(t,j=7.4hz,1h),7.49-7.37(m,4h),6.15(d,j=1.6hz,2h);13cnmr(100mhz,cdcl3)δ186.39,163.85,163.23,160.70,151.24,147.54,136.23,134.78,132.59,131.13(d,j=10.9hz),130.19,128.78,125.31,120.81,120.78,117.24,117.03,116.76,56.99(d,j=4.7hz);19fnmr(377mhz,cdcl3)δ-110.34;ir(kbr)3138,2359,1731,1682,1541,1402,1320,1195,1068,986,887,676cm-1;hrms(esi):m/z[m+h]+calcdforc18h13fno3:310.0874,found:310.0873.
实施例7
将7-氯-8-甲基喹啉(17.7mg,0.1mmol)、苯甲酰甲酸(30.0mg,0.2mmol)、三(二亚苄基丙酮)二钯(9.2mg,0.01mmol)、二乙酸碘苯(80.5mg,0.25mmol)、及一粒搅拌子放入反应管中,加入1毫升溶剂chcl3,密封反应管。将反应管置于70℃油浴反应锅中搅拌反应12小时。冷至室温后,用饱和碳酸氢钠溶液中和并用乙酸乙酯萃取。合并萃取液并用无水硫酸钠干燥、过滤、滤液经减压浓缩后,以乙酸乙酯:石油醚=1:4(体积比)为洗脱剂对粗产物进行柱层析得到纯品。黄色固体,熔点129-131℃,产率69%。1hnmr(400mhz,cdcl3)δ9.00(dd,j=4.0,1.6hz,1h),8.15(dd,j=8.2,1.4hz,1h),8.07-8.05(m,2h),7.81(d,j=8.8hz,1h),7.63-7.57(m,2h),7.49-7.44(m,3h),6.29(s,2h);13cnmr(100mhz,cdcl3)δ186.44,163.93,151.12,147.48,137.35,136.18,134.79,132.58,130.19,130.00,128.79,128.10,126.89,121.58,60.64;ir(kbr)3547,3140,1727,1687,1491,1402,1195,1128,982,833,796,684cm-1;hrms(esi):m/z[m+k]+calcdforc18h12clkno3:364.0137,found:364.0135.
实施例8
将6-溴-8-甲基喹啉(22.1mg,0.1mmol)、苯甲酰甲酸(30.0mg,0.2mmol)、三(二亚苄基丙酮)二钯(9.2mg,0.01mmol)、二乙酸碘苯(80.5mg,0.25mmol)、及一粒搅拌子放入反应管中,加入1毫升溶剂chcl3,密封反应管。将反应管置于70℃油浴反应锅中搅拌反应12小时。冷至室温后,用饱和碳酸氢钠溶液中和并用乙酸乙酯萃取。合并萃取液并用无水硫酸钠干燥、过滤、滤液经减压浓缩后,以乙酸乙酯:石油醚=1:4(体积比)为洗脱剂对粗产物进行柱层析得到纯品。黄色固体,熔点47-49℃,产率80%。1hnmr(400mhz,cdcl3)δ8.96-8.95(m,1h),8.08(t,j=8.8hz,3h),7.99-7.92(m,2h),7.66(t,j=7.6hz,1h),7.53-7.46(m,3h),6.10(s,2h);13cnmr(100mhz,cdcl3)δ186.14,163.60,150.27,144.60,135.30,135.07,132.41,132.02,130.45,130.22,129.34,128.95,122.41,120.13,63.61;ir(kbr)3136,2363,1731,1685,1654,1522,1491,1402,1197,989,868,785,675cm-1;hrms(esi):m/z[m+h]+calcdforc18h13brno3:370.0073,found:370.0069.
实施例9
将8-甲基-5-硝基喹啉(18.8mg,0.1mmol)、苯甲酰甲酸(30.0mg,0.2mmol)、三(二亚苄基丙酮)二钯(9.2mg,0.01mmol)、二乙酸碘苯(80.5mg,0.25mmol)、及一粒搅拌子放入反应管中,加入1毫升溶剂chcl3,密封反应管。将反应管置于70℃油浴反应锅中搅拌反应12小时。冷至室温后,用饱和碳酸氢钠溶液中和并用乙酸乙酯萃取。合并萃取液并用无水硫酸钠干燥、过滤、滤液经减压浓缩后,以乙酸乙酯:石油醚=1:4(体积比)为洗脱剂对粗产物进行柱层析得到纯品。黄色固体,熔点124-125℃、产率70%。1hnmr(400mhz,cdcl3)δ9.07-9.03(m,2h),8.39(d,j=8.0hz,1h),8.09-8.07(m,2h),7.94(d,j=8.0hz,1h),7.73-7.67(m,2h),7.54(t,j=7.8hz,2h),6.21(s,2h);13cnmr(100mhz,cdcl3)δ185.85,163.31,150.91,145.48,140.81,135.23,132.32,130.20,129.03,125.61,124.38,124.26,120.97,63.93;ir(kbr)3466,1843,1716,1694,1683,1519,1402,1338,1193,992,816,753,693cm-1;hrms(esi):m/z[m+h]+calcdforc18h13n2o5:337.0819,found:337.0817.
实施例10
将8-甲基-6-硝基喹啉(18.8mg,0.1mmol)、苯甲酰甲酸(30.0mg,0.2mmol)、三(二亚苄基丙酮)二钯(9.2mg,0.01mmol)、二乙酸碘苯(80.5mg,0.25mmol)、及一粒搅拌子放入反应管中,加入1毫升溶剂chcl3,密封反应管。将反应管置于70℃油浴反应锅中搅拌反应12小时。冷至室温后,用饱和碳酸氢钠溶液中和并用乙酸乙酯萃取。合并萃取液并用无水硫酸钠干燥、过滤、滤液经减压浓缩后,以乙酸乙酯:石油醚=1:4(体积比)为洗脱剂对粗产物进行柱层析得到纯品。黄色固体,熔点123-125℃,产率72%。1hnmr(400mhz,cdcl3)δ9.14(dd,j=4.2,1.8hz,1h),8.81(d,j=2.4hz,1h),8.62(d,j=2.4hz,1h),8.40(dd,j=8.4,1.6hz,1h),8.10-8.08(m,2h),7.70-7.63(m,2h),7.54(t,j=7.8hz,2h),6.18(s,2h);13cnmr(100mhz,cdcl3)δ185.83,163.45,153.29,147.83,145.26,138.16,135.91,135.13,132.40,130.18,129.00,127.13,124.84,123.30,121.68,63.52;ir(kbr)3545,2354,1716,1683,1651,1519,1399,1195,1176,992,978,740,693cm-1;hrms(esi):m/z[m+h]+calcdforc18h13n2o5:337.0819,found:337.0812.
实施例11
将8-乙基喹啉(15.7mg,0.1mmol)、苯甲酰甲酸(30.0mg,0.2mmol)、三(二亚苄基丙酮)二钯(9.2mg,0.01mmol)、二乙酸碘苯(80.5mg,0.25mmol)、及一粒搅拌子放入反应管中,加入1毫升溶剂chcl3,密封反应管。将反应管置于70℃油浴反应锅中搅拌反应12小时。冷至室温后,用饱和碳酸氢钠溶液中和并用乙酸乙酯萃取。合并萃取液并用无水硫酸钠干燥、过滤、滤液经减压浓缩后,以乙酸乙酯:石油醚=1:4(体积比)为洗脱剂对粗产物进行柱层析得到纯品。黄色油状,产率54%。1hnmr(400mhz,cdcl3)δ8.98(dd,j=4.2,1.8hz,1h),8.18(dd,j=8.2,1.8hz,1h),8.04-8.02(m,2h),7.86(d,j=6.8hz,1h),7.80(d,j=7.6hz,1h),7.64(t,j=7.6hz,1h),7.56(t,j=7.6hz,1h),7.51-7.44(m,4h),1.86(d,j=6.4hz,3h);13cnmr(100mhz,cdcl3)δ186.69,163.24,149.80,145.07,139.09,136.25,134.82,132.57,130.14,128.85,128.18,127.94,126.38,125.51,121.35,70.93,21.87;ir(kbr)3138,2932,1735,1687,1597,1317,1297,1200,1178,1070,984,829,796,687cm-1;hrms(esi):m/z[m+na]+calcdforc19h15nnao3:328.0944,found:328.0940.
实施例12
将8-苄基喹啉(21.9mg,0.1mmol)、苯甲酰甲酸(30.0mg,0.2mmol)、三(二亚苄基丙酮)二钯(9.2mg,0.01mmol)、二乙酸碘苯(80.5mg,0.25mmol)、及一粒搅拌子放入反应管中,加入1毫升溶剂chcl3,密封反应管。将反应管置于70℃油浴反应锅中搅拌反应12小时。冷至室温后,用饱和碳酸氢钠溶液中和并用乙酸乙酯萃取。合并萃取液并用无水硫酸钠干燥、过滤、滤液经减压浓缩后,以乙酸乙酯:石油醚=1:4(体积比)为洗脱剂对粗产物进行柱层析得到纯品。黄色油状,产率52%。1hnmr(400mhz,cdcl3)δ8.99(dd,j=4.4,1.6hz,1h),8.52(s,1h),8.16(dd,j=8.4,1.6hz,1h),8.00(d,j=7.2hz,2h),7.81(dd,j=11.6,7.6hz,2h),7.62(t,j=7.8hz,3h),7.54(t,j=7.8hz,1h),7.48-7.42(m,3h),7.37-7.28(m,3h);13cnmr(100mhz,cdcl3)δ186.43,162.99,150.01,139.27,137.52,136.14,134.85,132.53,130.17,128.84,128.34,128.25,128.08,127.39,127.29,126.30,121.46,74.42.ir(kbr)3137,2855,1733,1683,1597,1498,1452,1399,1288,1196,1174,986,820,799,698cm-1;hrms(esi):m/z[m+h]+calcdforc24h18no3:368.1281,found:368.1287.
实施例13
将8-甲基喹啉(14.3mg,0.1mmol)、2-氧代-2-(邻甲苯基)乙酸(32.8mg,0.2mmol)、三(二亚苄基丙酮)二钯(9.2mg,0.01mmol)、二乙酸碘苯(80.5mg,0.25mmol)、及一粒搅拌子放入反应管中,加入1毫升溶剂chcl3,密封反应管。将反应管置于70℃油浴反应锅中搅拌反应12小时。冷至室温后,用饱和碳酸氢钠溶液中和并用乙酸乙酯萃取。合并萃取液并用无水硫酸钠干燥、过滤、滤液经减压浓缩后,以乙酸乙酯:石油醚=1:4(体积比)为洗脱剂对粗产物进行柱层析得到纯品。黄色固体,熔点106-108℃,产率79%。1hnmr(400mhz,cdcl3)δ8.97(dd,j=4.2,1.8hz,1h),8.18(dd,j=8.2,1.8hz,1h),7.86-7.78(m,3h),7.56(dd,j=8.4,7.2hz,1h),7.48-7.44(m,2h),7.29-7.26(m,2h),6.13(s,2h),2.61(s,3h);13cnmr(100mhz,cdcl3)δ188.73,164.70,150.03,146.17,141.54,136.24,133.77,132.94,132.81,132.32,131.15,129.25,128.72,128.22,126.25,125.89,121.50,64.33,21.57;ir(kbr)3137,2361,1716,1687,1597,1571,1402,1314,1260,1135,1074,964,829,675cm-1;hrms(esi):m/z[m+na]+calcdforc19h15nnao3:328.0944,found:328.0940.
实施例14
将8-甲基喹啉(14.3mg,0.1mmol)、2-氧代-2-(对甲苯基)乙酸(32.8mg,0.2mmol)、三(二亚苄基丙酮)二钯(9.2mg,0.01mmol)、二乙酸碘苯(80.5mg,0.25mmol)、及一粒搅拌子放入反应管中,加入1毫升溶剂chcl3,密封反应管。将反应管置于70℃油浴反应锅中搅拌反应12小时。冷至室温后,用饱和碳酸氢钠溶液中和并用乙酸乙酯萃取。合并萃取液并用无水硫酸钠干燥、过滤、滤液经减压浓缩后,以乙酸乙酯:石油醚=1:4(体积比)为洗脱剂对粗产物进行柱层析得到纯品。黄色油状,产率72%。1hnmr(400mhz,cdcl3)δ8.97(dd,j=4.0,1.6hz,1h),8.18(dd,j=8.4,1.6hz,1h),7.95(d,j=8.0hz,2h),7.84(t,j=6.8hz,2h),7.56(t,j=7.6hz,1h),7.46(dd,j=8.2,4.2hz,1h),7.27(d,j=8.4hz,2h),6.14(s,2h),2.42(s,3h);13cnmr(100mhz,cdcl3)δ186.12,164.10,150.01,146.19,146.15,136.25,132.97,130.33,130.14,129.58,129.16,128.68,128.23,126.25,121.49,64.31,21.92;ir(kbr)3152,1735,1685,1605,1500,1399,1305,1198,1172,997,828,792,760cm-1;hrms(esi):m/z[m+h]+calcdforc19h16no3:306.1125,found:306.1126.
实施例15
将8-甲基喹啉(14.3mg,0.1mmol)、2-(4-甲氧基苯基)-2-氧代乙酸(36.0mg,0.2mmol)、三(二亚苄基丙酮)二钯(9.2mg,0.01mmol)、二乙酸碘苯(80.5mg,0.25mmol)、及一粒搅拌子放入反应管中,加入1毫升溶剂chcl3,密封反应管。将反应管置于70℃油浴反应锅中搅拌反应12小时。冷至室温后,用饱和碳酸氢钠溶液中和并用乙酸乙酯萃取。合并萃取液并用无水硫酸钠干燥、过滤、滤液经减压浓缩后,以乙酸乙酯:石油醚=1:4(体积比)为洗脱剂对粗产物进行柱层析得到纯品。黄色油状,产率75%。1hnmr(400mhz,cdcl3)δ8.96(dd,j=4.2,1.4hz,1h),8.18(dd,j=8.4,1.2hz,1h),8.04(d,j=8.8hz,2h),7.84(t,j=8.2hz,2h),7.55(t,j=7.6hz,1h),7.45(dd,j=8.2,4.2hz,1h),6.94(d,j=8.8hz,2h),6.13(s,2h),3.87(s,3h);13cnmr(100mhz,cdcl3)δ184.94,165.00,164.23,150.00,146.13,136.27,132.98,132.75,129.13,128.67,128.22,126.26,125.61,121.49,114.21,64.26,55.65;ir(kbr)3146,2929,1737,1603,1502,1401,1252,1162,1028,827,790,604cm-1;hrms(esi):m/z[m+h]+calcdforc19h16no4:322.1074,found:322.1068.
实施例16
将8-甲基喹啉(14.3mg,0.1mmol)、2-(2-氟苯基)-2-氧代乙酸(33.6mg,0.2mmol)、三(二亚苄基丙酮)二钯(9.2mg,0.01mmol)、二乙酸碘苯(80.5mg,0.25mmol)、及一粒搅拌子放入反应管中,加入1毫升溶剂chcl3,密封反应管。将反应管置于70℃油浴反应锅中搅拌反应12小时。冷至室温后,用饱和碳酸氢钠溶液中和并用乙酸乙酯萃取。合并萃取液并用无水硫酸钠干燥、过滤、滤液经减压浓缩后,以乙酸乙酯:石油醚=1:4(体积比)为洗脱剂对粗产物进行柱层析得到纯品。黄色固体,熔点86-88℃,产率74%。1hnmr(400mhz,cdcl3)δ8.96(dd,j=4.2,1.8hz,1h),8.18(dd,j=8.2,1.8hz,1h),7.94(td,j=7.6,1.6hz,1h),7.84(dd,j=11.2,7.6hz,2h),7.64-7.54(m,2h),7.45(dd,j=8.2,4.2hz,1h),7.30-7.26(m,1h),7.15-7.11(m,1h),6.16(s,2h);13cnmr(100mhz,cdcl3)δ183.92,164.05,161.48,150.00,146.04,136.76,136.67,136.26,132.82,131.05,131.04,129.01,128.57,128.17,126.26,124.81,124.78,121.78(d,j=10.2hz),121.44,116.64(d,j=21.3hz),64.49;19fnmr(377mhz,cdcl3)δ-110.80;ir(kbr)3138,1744,1688,1610,1484,1402,1312,1259,1193,1103,992,828,790,656cm-1;hrms(esi):m/z[m+na]+calcdforc18h12fnnao3:332.0693,found:332.0681.
实施例17
将8-甲基喹啉(14.3mg,0.1mmol)、2-(4-氯苯基)-2-氧代乙酸(36.8mg,0.2mmol)、三(二亚苄基丙酮)二钯(9.2mg,0.01mmol)、二乙酸碘苯(80.5mg,0.25mmol)、及一粒搅拌子放入反应管中,加入1毫升溶剂chcl3,密封反应管。将反应管置于70℃油浴反应锅中搅拌反应12小时。冷至室温后,用饱和碳酸氢钠溶液中和并用乙酸乙酯萃取。合并萃取液并用无水硫酸钠干燥、过滤、滤液经减压浓缩后,以乙酸乙酯:石油醚=1:4(体积比)为洗脱剂对粗产物进行柱层析得到纯品。黄色固体,熔点112-114℃,产率76%。1hnmr(400mhz,cdcl3)δ8.97(dd,j=4.2,1.8hz,1h),8.19(dd,j=8.2,1.8hz,1h),8.03-8.01(m,2h),7.85(d,j=7.6hz,2h),7.56(t,j=7.8hz,1h),7.48-7.43(m,3h),6.13(s,2h);13cnmr(100mhz,cdcl3)δ185.01,163.33,150.07,146.21,141.57,136.29,132.71,131.58,131.01,129.48,129.24,128.92,128.27,126.24,121.56,64.65;ir(kbr)3140,2367,1710,1687,1655,1588,1402,1308,1196,1174,1036,993,849,792cm-1;hrms(esi):m/z[m+h]+calcdforc18h13clno3:326.0579,found:326.0578.
实施例18
将8-甲基喹啉(14.3mg,0.1mmol)、2-(3-氯苯基)-2-氧代乙酸(36.8mg,0.2mmol)、三(二亚苄基丙酮)二钯(9.2mg,0.01mmol)、二乙酸碘苯(80.5mg,0.25mmol)、及一粒搅拌子放入反应管中,加入1毫升溶剂chcl3,密封反应管。将反应管置于70℃油浴反应锅中搅拌反应12小时。冷至室温后,用饱和碳酸氢钠溶液中和并用乙酸乙酯萃取。合并萃取液并用无水硫酸钠干燥、过滤、滤液经减压浓缩后,以乙酸乙酯:石油醚=1:4(体积比)为洗脱剂对粗产物进行柱层析得到纯品。白色固体,熔点91-93℃,产率78%。1hnmr(400mhz,cdcl3)δ9.01(d,j=4.0hz,1h),8.20(d,j=8.4hz,1h),8.04(s,1h),7.96(d,j=8.0hz,1h),7.88-7.85(m,2h),7.61-7.55(m,2h),7.49-7.40(m,2h),6.13(s,2h);13cnmr(100mhz,cdcl3)δ185.05,163.15,150.23,146.25,136.27,135.16,134.75,134.17,132.63,130.12,129.65,129.01,128.28,126.21,121.57,64.78;ir(kbr)3138,1733,1684,1573,1498,1401,1312,1192,1077,1021,895,783,697cm-1;hrms(esi):m/z[m+h]+calcdforc18h13clno3:326.0579,found:326.0575.
实施例19
将8-甲基喹啉(14.3mg,0.1mmol)、2-(2-溴苯基)-2-氧代乙酸(45.6mg,0.2mmol)、三(二亚苄基丙酮)二钯(9.2mg,0.01mmol)、二乙酸碘苯(80.5mg,0.25mmol)、及一粒搅拌子放入反应管中,加入1毫升溶剂chcl3,密封反应管。将反应管置于70℃油浴反应锅中搅拌反应12小时。冷至室温后,用饱和碳酸氢钠溶液中和并用乙酸乙酯萃取。合并萃取液并用无水硫酸钠干燥、过滤、滤液经减压浓缩后,以乙酸乙酯:石油醚=1:4(体积比)为洗脱剂对粗产物进行柱层析得到纯品。黄色油状,产率72%。1hnmr(400mhz,cdcl3)δ8.95-8.94(m,1h),8.17(d,j=8.4hz,1h),7.84(t,j=8.8hz,2h),7.73(dd,j=7.2,1.6hz,1h),7.61(d,j=6.8hz,1h),7.55(t,j=7.6hz,1h),7.46-7.40(m,3h),6.14(s,2h);13cnmr(100mhz,cdcl3)δ187.06,162.58,150.00,146.08,136.22,135.30,134.07,133.86,132.69,132.10,129.08,128.59,128.16,127.63,126.22,121.85,121.46,67.88;ir(kbr)3118,1942,1732,1694,1502,1435,1399,1057,987,829,792,667cm-1;hrms(esi):m/z[m+h]+calcdforc18h13brno3:370.0073,found:370.0080.
实施例20
将8-甲基喹啉(14.3mg,0.1mmol)、2-(4-溴苯基)-2-氧代乙酸(45.6mg,0.2mmol)、三(二亚苄基丙酮)二钯(9.2mg,0.01mmol)、二乙酸碘苯(80.5mg,0.25mmol)、及一粒搅拌子放入反应管中,加入1毫升溶剂chcl3,密封反应管。将反应管置于70℃油浴反应锅中搅拌反应12小时。冷至室温后,用饱和碳酸氢钠溶液中和并用乙酸乙酯萃取。合并萃取液并用无水硫酸钠干燥、过滤、滤液经减压浓缩后,以乙酸乙酯:石油醚=1:4(体积比)为洗脱剂对粗产物进行柱层析得到纯品。白色固体,熔点125-127℃,产率77%。1hnmr(400mhz,cdcl3)δ8.98(d,j=3.6hz,1h),8.20(d,j=8.4hz,1h),7.94(d,j=8.4hz,2h),7.85(d,j=8.0hz,2h),7.63-7.55(m,3h),7.48(dd,j=8.2,4.2hz,1h),6.12(s,2h);13cnmr(100mhz,cdcl3)δ185.25,163.30,150.09,146.19,136.32,132.65,132.24,131.61,131.39,130.51,129.54,128.96,128.28,126.25,121.58,64.69;ir(kbr)3146,1737,1685,1584,1498,1402,1312,1267,1193,1069,992,827,790cm-1;hrms(esi):m/z[m+k]+calcdforc18h12brkno3:377.1485,found:377.1475.
实施例21
将8-甲基喹啉(14.3mg,0.1mmol)、2-(萘-1-基)-2-氧代乙酸(40.0mg,0.2mmol)、三(二亚苄基丙酮)二钯(9.2mg,0.01mmol)、二乙酸碘苯(80.5mg,0.25mmol)、及一粒搅拌子放入反应管中,加入1毫升溶剂chcl3,密封反应管。将反应管置于70℃油浴反应锅中搅拌反应12小时。冷至室温后,用饱和碳酸氢钠溶液中和并用乙酸乙酯萃取。合并萃取液并用无水硫酸钠干燥、过滤、滤液经减压浓缩后,以乙酸乙酯:石油醚=1:4(体积比)为洗脱剂对粗产物进行柱层析得到纯品。黄色固体,熔点92-94℃,产率71%。1hnmr(400mhz,cdcl3)δ9.07(d,j=8.4hz,1h),8.98(dd,j=4.4,1.6hz,1h),8.17(dd,j=8.2,1.4hz,1h),8.12-8.07(m,2h),7.89-7.82(m,3h),7.67-7.63(m,1h),7.58-7.53(m,2h),7.51-7.44(m,2h),6.17(s,2h);13cnmr(100mhz,cdcl3)δ188.85,164.67,150.05,146.18,136.30,135.88,134.63,133.89,132.93,131.02,129.38,129.27,128.79,128.73,128.25,128.10,127.00,126.27,125.68,124.34,121.52,64.49;ir(kbr)3138,1737,1677,1573,1506,1402,1301,1215,1174,1092,951,828,779cm-1;hrms(esi):m/z[m+k]+calcdforc22h15kno3:380.0684,found:380.0672.
实施例22
将8-甲基喹啉(14.3mg,0.1mmol)、2-氧代-2-(噻吩-2-基)乙酸(31.2mg,0.2mmol)、三(二亚苄基丙酮)二钯(9.2mg,0.01mmol)、二乙酸碘苯(80.5mg,0.25mmol)、及一粒搅拌子放入反应管中,加入1毫升溶剂chcl3,密封反应管。将反应管置于70℃油浴反应锅中搅拌反应12小时。冷至室温后,用饱和碳酸氢钠溶液中和并用乙酸乙酯萃取。合并萃取液并用无水硫酸钠干燥、过滤、滤液经减压浓缩后,以乙酸乙酯:石油醚=1:4(体积比)为洗脱剂对粗产物进行柱层析得到纯品。黄色油状,产率55%。1hnmr(400mhz,cdcl3)δ8.95(dd,j=4.0,1.6hz,1h),8.19-8.15(m,2h),7.86-7.78(m,3h),7.55(t,j=7.8hz,1h),7.46(dd,j=8.2,4.2hz,1h),7.16(t,j=4.4hz,1h),6.12(s,2h);13cnmr(100mhz,cdcl3)δ176.55,161.72,150.05,146.14,139.36,137.47,137.39,136.26,132.81,129.12,128.69,128.68,128.23,126.26,121.51,64.83;ir(kbr)3118,1735,1666,1597,1582,1502,1403,1299,1191,1083,1058,954,827,732cm-1;hrms(esi):m/z[m+h]+calcdforc16h12no3s:298.0532,found:298.0533.
对照试验组1~29:
将8-甲基喹啉(14.3mg,0.1mmol)、苯甲酰甲酸(30.0mg,0.2mmol)、钯催化剂、氧化剂、1毫升溶剂及一粒搅拌子放入反应管中,密封反应管。加热反应,产物采用1hnmr定量分析;各对照试验组的具体反应条件如表1所示。
表18-甲基喹啉与苯甲酰甲酸的脱氢偶联反应对照组实验
从上表可以看出,催化剂的种类和用量对该反应有较大影响,用量偏低,产率也较低;但对于该底物最好0.1毫摩尔三(二亚苄基丙酮)二钯。
从上表可以看出,氧化剂的种类和用量对该反应有较大影响,用量偏低,产率也较低;但对于该底物最好0.25毫摩尔醋酸碘苯。
从上表可以看出,溶剂的类型及用量对该反应也有较大影响,反应效果最好的溶剂是三氯甲烷。
从上表还可以看出,该反应在10小时可顺利进行。反应12小时即可以达到最高产率、而进一步延长反应时间、产率变化不大。
对照试验组30:
将8-甲基喹啉(1g,7mmol)、苯甲酰甲酸(2.1g,14mmol)、三(二亚苄基丙酮)二钯(64.0mg,0.07mmol)、二乙酸碘苯(5.6g,0.25mmol)、及一粒搅拌子放入反应管中,加入7毫升溶剂chcl3,密封反应管。将反应管置于70℃油浴反应锅中搅拌反应48小时。冷至室温后,用饱和碳酸氢钠溶液中和并用乙酸乙酯萃取。合并萃取液并用无水硫酸钠干燥,过滤,滤液经减压浓缩后,以乙酸乙酯:石油醚=1:4(体积比)为洗脱剂对粗产物进行柱层析得到纯品1.6g,产率78%。
由此可见、该反应可以在低钯催化剂负载量(1mol%)下容易地扩增至克水平、具有好的工业化应用前景。
- 还没有人留言评论。精彩留言会获得点赞!