氧化荷苞牡丹碱的合成方法与流程
本发明涉及氧化荷苞牡丹碱的合成方法,属于医药技术领域。
背景技术:
荷苞牡丹碱主要来源于罂粟科植物水上叶荷苞牡丹(dicentraspectabilis),防已科植物荷苞地不容(stephaniadicentrinifera),别名痛可宁或山乌龟碱。有研究学者通过分离的方法,从o.leucoxylon中分离出具有生物活性的阿朴啡、异喹啉和双异喹啉生物碱,这些化合物包含的荷苞牡丹碱,具有抗肿瘤、镇痛镇静作用及抗菌的生物活性。s-荷苞牡丹碱可以从黑壳楠(linderamegaphylla(lauraceae))中分离得到,通过对它进行的抗肿瘤研究发现,其对人类肝细胞瘤huh-7有较显著的抗肿瘤活性。s-荷苞牡丹碱在对来自七种不同组织的二十一种人类肿瘤细胞的体外活性研究中,其对所有的肿瘤细胞均表现出不同程度的抗肿瘤活性,ic50值范围由对食道癌细胞hce-6的0.4μm到对肝细胞瘤ha22t的29μm,实验证明,s-荷苞牡丹碱对各种癌细胞具有一定的抑制活性(huangrl,chencc,huangyl,oujc,hucp,chencf,changc.anti-tumoreffectsofd-dicentrinefromtherootoflinderamegaphyll.plantamed,1998,64(3),212-215.)。例如肾上腺素受体作用的活性,离子通道作用活性,细胞毒性,抗氧化活性,抗血小板聚集活性和抗锥虫活性等。
氧化的荷苞牡丹碱(dicentrinone,即氧化荷苞牡丹碱,简称dco),具有杀利什曼(原)虫及锥虫的活性,dco对trypanocidal、antileishmanial以及rad52修复缺失型的rs322酵母菌株具有较好的抑制活性。同时,氧化荷苞牡丹碱是拓朴异构酶ⅰ、ⅱ的抑制剂,且其对细胞的毒性也比较弱(zhoub,johnsonrk,matternmr,wangx,hechtsm.isolationandbiochemicalcharacterizationofanewtopoisomeraseiinhibitorfromocotealeucoxylon.j.nat.prod.2000,63,217–221.stevignyc,baillyc,quetin-leclercq.cytotoxicandantitumorpotentialitiesofaporphinoidalkaloids.curr.med.chem.-anti-canceragents,2005,5(2),173-182(10).),很有希望成为极具潜力的抗癌新药。但由于dco生物碱在自然界含量极低,很难满足构效关系和活性药理研究等需要,因此,为了充分开发dco在医药方面的潜在作用,通过有机合成的方法得到dco是十分重要的。
技术实现要素:
本发明要解决的技术问题是提供一种产率高的氧化荷苞牡丹碱的合成方法。
本发明所述的氧化荷苞牡丹碱的合成方法,是以3,4-二甲氧基苯乙酸为起始原料,按以下合成路线进行合成:
其中,所述的还原剂为选自硼氢化钠、硼氢化锂和硼氢化钾中的一种或两种以上的组合。
上述氧化荷苞牡丹碱更为具体的合成方法包括以下步骤:
1)化合物(2)的合成:
取3,4-二甲氧基苯乙酸溶于冰醋酸中,加入溴素进行反应,所得反应物倒入冰水中,静置、抽滤,得到化合物(2);
2)化合物(3)的合成:
取化合物(2)置于二氯亚砜中,于加热或不加热条件下反应,反应物蒸除未反应的二氯亚砜,得到淡黄色液体;取胡椒乙胺溶于第一有机溶剂中,所得溶液加入到前述淡黄色液体中,于加热或不加热条件下反应,反应物蒸除溶剂,得到化合物(3);
3)化合物(4)的合成:
取化合物(3)置于第一有机溶剂中,加入三氯氧磷,于加热或不加热条件下反应,反应物蒸除溶剂,得到化合物(4);
4)化合物(5)的合成:
取化合物(4)溶于第一有机溶剂中,加入过量的还原剂,于加热或不加热条件下反应,向所得反应物中加入稀酸以除去未反应的还原剂,所得物料用萃取剂进行萃取,收集有机相,用饱和碳酸氢钠溶液洗涤,干燥,得到化合物(5);
5)化合物(6)的合成:
取化合物(5)溶于第一有机溶剂中,调节体系的ph值至碱性,加入二碳酸二叔丁酯,于加热或不加热条件下反应,所得反应物用萃取剂进行萃取,收集有机相,旋干,得到化合物(6);
6)化合物(7)的合成:
取三环己基磷和醋酸钯溶于第二有机溶剂中,调节体系的ph=8-10,向其中加入化合物(6),在气氛保护且加热条件下反应,所得反应物用酸中和后再用萃取剂进行萃取,收集有机相,用饱和碳酸氢钠溶液洗涤,干燥,得到化合物(7);
7)化合物(8)的合成:
取化合物(7)溶于四氢呋喃中,冰浴条件下加入四氢铝锂,在气氛保护条件下反应,调节所得反应物的ph=8-9,抽滤,收集滤液,滤液旋干,得到化合物(8);
8)氧化荷苞牡丹碱即化合物(9)的合成:
取化合物(8)溶于冰醋酸中,加入乙酸锰(ⅲ)(c6h9mno6.2(h2o)),于加热或不加热条件下反应,所得反应物过滤,收集滤液,蒸除溶剂,得到氧化荷苞牡丹碱粗品;
上述合成方法中,所述的第一有机溶剂为选自氯仿、二氯甲烷、甲醇、乙醇、丙醇和正丁醇中的一种或两种以上的组合;所述的第二有机溶剂为n,n-二甲基甲酰胺(dma)和/或n,n-二甲基乙酰胺(dmac);所述的萃取剂为选自氯仿、二氯甲烷和乙酸乙酯中的一种。
上述合成方法中:
步骤1)中,所述3,4-二甲氧基苯乙酸和溴素的摩尔比为化学计量比,在实际操作中通常选取3,4-二甲氧基苯乙酸和溴素的摩尔比为1:1.2-1:1.5;反应优选在常温下进行,反应是否完全可采用薄层层析跟踪检测,在上述限定条件下,反应至完全大约需要1-3h。该步骤中,冰醋酸的用量可根据需要确定,通常情况下,以0.1mol的3,4-二甲氧基苯乙酸为基准,全部原料优选用180-250ml的冰醋酸来溶解。优选将溴素先用冰醋酸溶解后再加入到3,4-二甲氧基苯乙酸的冰醋酸溶液中进行反应。本步骤的产率在95%以上。
步骤2)中,二氯亚砜既做溶剂也是反应物,化合物(2)和二氯亚砜的摩尔比通常为1:1.5-2.0;所述化合物(2)和二氯亚砜优选是在加热条件下进行,更优选在60至溶剂的回流温度条件下进行回流反应,反应是否完全可采用薄层层析跟踪检测,在上述限定条件下,反应至完全大约需要3-5h。本步骤的产率在80%以上。
步骤3)中,化合物(3)和三氯氧磷的摩尔比为化学计量比,在实际操作中通常选取化合物(3)和三氯氧磷的摩尔比为1:4-1:5;所述化合物(3)和三氯氧磷优选是在加热条件下进行,更优选在60℃至溶剂的回流温度条件下进行回流反应,反应是否完全可采用薄层层析跟踪检测,在上述限定条件下,反应至完全大约需要2-4h。本步骤的产率在80%以上。
步骤4)中,所述的反应优选在常温条件下进行,反应至完全大约需要8-12h。所述的稀酸通常为0.5-2mol/l的盐酸溶液。本步骤的产率在85%以上。
步骤5)中,化合物(5)和二碳酸二叔丁酯(boc酸酐)的摩尔比为化学计量比,在实际操作中通常选取化合物(5)和二碳酸二叔丁酯的摩尔比为1:1.0-1:1.5;反应优选在常温条件下进行,反应至完全大约需要3-5h。本步骤的产率在95%以上。
步骤6)中,化合物(6)和三环己基磷的摩尔比为化学计量比,在实际操作中通常选取化合物(6)和三环己基磷的摩尔比为1:1-1:1.2;反应更优选是在120-135℃条件下进行,在上述限定条件下,反应至完全大约需要20-30h。该步骤中,醋酸钯作催化剂使用,其用量优选为化合物(6)质量的2%以上,更优选为化合物(6)质量的2.5-3%。本步骤的产率在82%以上。
步骤5)、6)和7)中,通常采用现有常用的碱性物质的水溶液来调节体系的ph值,优选采用碳酸钾水溶液、氢氧化钠水溶液、氨水或三乙胺等进行调节。步骤5)中,优选调节体系的ph≥8,更优选为调节体系的ph=8.5-10。
步骤7)中,化合物(7)和四氢铝锂的摩尔比为化学计量比,在实际操作中通常选取化合物(7)和四氢铝锂的摩尔比为1:8-1:10;反应更优选是在加热条件下进行,更优选是在60-70℃条件下回流反应,在上述限定条件下,反应至完全大约需要20-30h。本步骤的产率在90%以上。
步骤8)中,乙酸锰(ⅲ)作催化剂使用,其用量优选为化合物(8)质量的2倍以上,更优选为化合物(8)质量的3-4倍。所述化合物(8)和乙酸锰(ⅲ)的反应优选是在加热条件下进行,更优选是在60-70℃条件下回流反应,在上述限定条件下,反应至完全大约需要4-6h。本步骤的产率在50%以上。
在上述步骤1)-7)中,所得的化合物均为粗品,为了减少引入到后续步骤的杂质,同时提高目标物粗品的纯度,优选是将所得化合物进行纯化后再用于后续操作。所述的纯化具体可以是将所得化合物粗品用溶剂进行重结晶后再用于后续操作,进行重结晶的溶剂可以是甲醇和/或乙醇,或者是水与甲醇或乙醇按1:2-3的体积比组成的混合物。
同理,为了提高氧化荷苞牡丹碱的纯度,优选在得到氧化荷苞牡丹碱粗品之后再进行纯化步骤,具体的纯化步骤为:将氧化荷苞牡丹碱粗品上硅胶柱层析,用由氯仿和甲醇,或者是由二氯甲烷和甲醇按30-35:1的体积比组成的混合溶剂洗脱,洗脱液蒸干溶剂,即得氧化荷苞牡丹碱。通过限定洗脱剂的比例,快速且准确地将目标物洗脱下来。
与现有技术相比,本发明提供了一种新的通过全合成制备氧化荷苞牡丹碱的方法,该方法合成路线简单易操作,且产率高(以纯化后的目标物计,产率在45%以上)。
附图说明
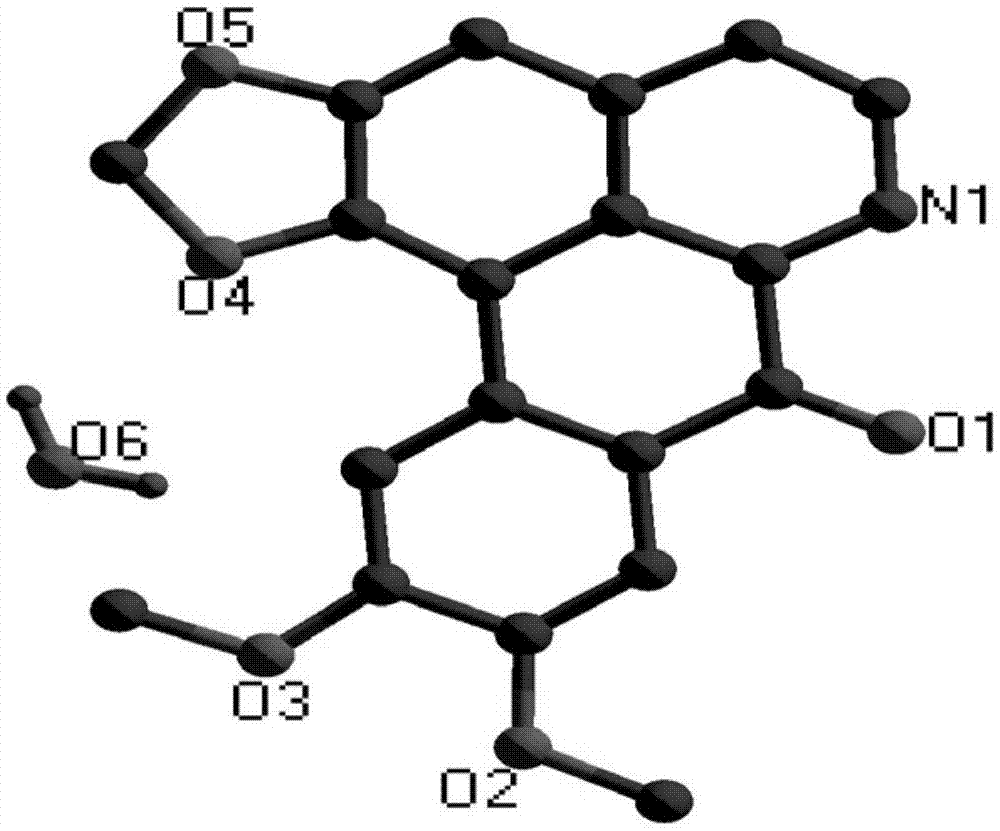
图1为本发明实施例1制得的最终产物的晶体结构图(去掉h原子);
图2为本发明所述氧化荷苞牡丹碱与dna作用的凝胶电泳图。
具体实施方式
下面结合具体实施例对本发明作进一步的详述,以更好地理解本发明的内容,但本发明并不限于以下实施例。
实施例1
1)化合物(2)的合成:
将72g3,4-二甲氧基苯乙酸溶解于600ml的冰醋酸中,常温下搅拌1h后加入7.2g溴素的冰醋酸(60ml)溶液,继续反应2h后,加入200ml冰水,有白色沉淀生成,过滤,滤饼用甲醇重结晶后,得到96g化合物(2),产率约为95%。
化合物(2)为白色固体,esi-msm/z273.02[(2)-h]-,13c-nmr(500mhz,dmso)δ:41.0816(c-2),56.2538(c-5),56.4330(c-6),114.9042(c-9),115.7982(c-3),115.9616(c-8),127.4302(c-10),148.5825(c-4),148.9687(c-8),172.0429(c-1),1h-nmr(500mhz,dmso)δ:3.6021(2h,s,h-2),3.7120(3h,s,h-5),3.7346(3h,s,h-6),6.9880(1h,s,h-3),7.0886(1h,s,h-8)。
2)化合物(3)的合成:
将100g化合物(2)溶于100ml的二氯亚砜中,76℃条件下回流1.5h,减压蒸馏掉没有反应完的二氯亚砜,得到淡黄色的液体,用400ml二氯甲烷溶解75g胡椒乙胺,并将其缓慢加入上述淡黄色液体中,常温搅拌4小时,减压蒸除二氯甲烷,用甲醇重结晶,约得到100g白色固体,产率约为80%。
化合物(3)为白色固体,esi-msm/z421.96[(3)+h]+;1h-nmr(500mhz,cdcl3)δ:2.485(2h,s,h-8),3.269(2h,s,h-12),3.408(2h,s,h-9),2.485(2h,s,h-8),3.697(6h,s,h-19,20),6.332(1h,s,h-15),6.374(1h,s,h-4),6.490(1h,s,h-7),6.611(1h,s,h-5),6.831(h,s,h-7)。13c-nmr(500mhz,cdcl3)δ:34.828(c-12),40.487(c-8),43.350(c-9),55.884(c-19,c-20),100.5927(c-1),107.965(c-4),108.704(c-5),113.524(c-14),114.470(c-15),115.434(c-16),121.286(c-7),126.336(c-13),132.033(c-6),145.849(c-3),147.468(c-17),148.525(c-2),148.745(c-18),169.440(c-11)。
3)化合物(4)的合成:
将100g的化合物(3)溶于1250ml氯仿中,然后加入180ml三氯氧磷(pocl3),回流反应3h,反应完成后减压蒸馏除溶剂,用饱和碳酸氢钠溶液洗涤,干燥,得到未纯化的化合物(4),产率约80%。
4)化合物(5)的合成:
将70g化合物(4)溶于300ml甲醇中,加入过量的硼氢化钠(80g),常温搅拌反应12h,缓慢加入用1mol/l的稀盐酸溶液以反应过量的硼氢化钠,所得物料用乙酸乙酯萃取,有机相用饱和碳酸氢钠溶液洗涤,再用甲醇重结晶,得到约78g化合物(5),产率约82%。
5)化合物(6)的合成:
将50g的化合物(5)溶于375ml氯仿中,然后加入150ml浓度为2mol/l的氢氧化钠水溶液,搅拌0.5h后(此时体系的ph=9),缓慢加入等物质的量的boc酸酐,继续搅拌4h,用氯仿萃取,有机相再用饱和碳酸氢钠溶液洗涤,之后减压蒸馏,得到57g化合物(6),产率约为96%。
6)化合物(7)的合成:
将0.32g的三环己基磷、3.15g碳酸钾和0.125g的醋酸钯溶于80ml干燥的dmf中(此时体系的ph=9),然后加入5g化合物(6),在惰性气体(氦气)保护下,于135℃回流24h,然后冷却,用1mol/l的盐酸中和后,用氯仿萃取,饱和碳酸氢钠溶液洗涤,干燥,用乙醇重结晶后得到4.3g化合物(7),产率约为92%。
7)化合物(8)的合成:
将5g的化合物(7)溶于130ml干燥的四氢呋喃中,在冰浴中缓慢加入4.4g的氢化铝锂,在惰性气体(氦气)保护下,50℃中回流24h,用稀氨水调节体系的ph=8,趁热抽滤,收集滤液,减压蒸馏除去溶剂,所得残余物用乙醇重结晶,析出固体后抽滤,得到化合物(8)约3.5g,产率约为90%。
8)氧化荷苞牡丹碱即化合物(9)的合成:
取2g化合物(8)溶于50ml的冰醋酸中,然后加入6.5g乙酸锰(ⅲ),于70℃条件下回流5h,反应物抽滤,滤液旋蒸除去溶剂,残余物即为氧化荷苞牡丹碱粗品。所得粗品用氯仿溶解,之后用饱和碳酸氢钠溶液洗涤三次,然后上硅胶柱层析(洗去残留的催化剂),用由氯仿和甲醇按30-32:1的体积比组成的混合溶剂洗脱,洗脱液蒸干溶剂,即得高纯度的黄色粉末产物1g,产率约50%。
取本实施例所得产物溶于由氯仿和甲醇按3:1的体积比组成的混合溶剂中,室温下缓慢挥发,在第15天时发现有明黄色棒状晶体出现,挑选合适的单晶进行结构表征:
1)红外表征:
用perkin-klmer公司的spectrumtwoft-irspectrometer傅立叶变换红外光谱仪(kbr压片),对本实施例制得的产物进行红外分析,所得红外光谱数据如下:
ir(kbrcm-1)(ar-h)3377(m),(-ch2-,v)2923,2851(m),(c=o)1638(vs),(c=c)1598,1572,1515,1424(s),(-ch2-,d)1459,(c-o)1279,1248,(c-n)1057,(-ch2-)776cm-1。
2)x-射线衍射分析:
取大小合适的单晶置于brukersmartaapex2ccd面探单晶衍射仪上,以石墨单色器单色化的mo-kα射线
所得晶体的结构图如图1所示,其晶体结构参数和部分键长、键角数据分别如下述表1和表2所示。
表1:
表2:
因此,可确定本实施例所得产物为目标产物氧化荷苞牡丹碱。
实施例2
重复实施例1,不同的是:
步骤3)-5)中,涉及的第一有机溶剂均改为乙醇;
步骤4)中,还原剂改为硼氢化锂;
步骤5)中,调节体系的ph=8,萃取剂改用氯仿;
步骤6)中,调节体系的ph=10,醋酸钯的用量改为化合物(6)质量的3%,第二有机溶剂改为dmac,萃取剂改用二氯甲烷;
步骤7)中,调节体系的ph=9;
步骤8)中,乙酸锰(ⅲ)的用量改为化合物(8)质量的4倍,上硅胶柱洗脱时的洗脱剂为由二氯甲烷和甲醇按32-35:1的体积比组成的混合溶剂洗脱。
将本实施例所得产物溶于由氯仿和甲醇按3:1的体积比组成的混合溶剂中,室温下缓慢挥发,将析出的晶体进行红外表征和单晶衍射分析,确定为目标产物氧化荷苞牡丹碱。
实施例3
重复实施例1,不同的是:
步骤3)中,涉及的第一有机溶剂改为正丁醇;
步骤4)中,涉及的第一有机溶剂改为正丙醇,还原剂改为硼氢化钾;
步骤5)中,涉及的第一有机溶剂改为甲醇,调节体系的ph=10;
步骤6)中,调节体系的ph=10,醋酸钯的用量改为化合物(6)质量的2%,第二有机溶剂改为dmac,萃取剂改用乙酸乙酯;
步骤7)中,调节体系的ph=8.5;
步骤8)中,乙酸锰(ⅲ)的用量改为化合物(8)质量的2倍,上硅胶柱洗脱时的洗脱剂为由二氯甲烷和甲醇按30:1的体积比组成的混合溶剂洗脱;
步骤1)-7)中,涉及重结晶操作时用的溶剂均改为乙醇和水按2:1的体积比的组合物。
将本实施例所得产物溶于由氯仿和甲醇按3:1的体积比组成的混合溶剂中,室温下缓慢挥发,将析出的晶体进行红外表征和单晶衍射分析,确定为目标产物氧化荷苞牡丹碱。
实验例1:氧化荷苞牡丹碱对多种人肿瘤细胞株的增殖抑制活性实验
本实验选用的细胞株为:膀胱癌细胞株t-24、人肝癌细胞hepg2、卵巢癌skov-3、卵巢癌耐药株细胞skov-3-ddp。用mtt法评价药物对活细胞生长及增殖的影响。
在初筛试验中,取处于对数生长期的系列肿瘤细胞株,用含10%新生牛血清的培养液配成单个细胞悬液,以每孔190μl(约1×104个/孔)细胞接种在96孔板,培养12h待细胞贴壁后,每孔分别加入不同浓度的样品10μl,每个梯度平行设4个复孔,其中的dmso是助溶剂,最终浓度不超过1%,同时设相应的阴性对照组(培养液中只有细胞和等量dmso,无药物)与空白对照组(培养液中只有等量的药物,无细胞),每个梯度也平行设4个复孔,药物作用时间为48小时。培养结束前4小时每孔加入10μlmtt(5mg/mlpbs),继续培养4小时后,吸弃上清液,再加入dmso100μl/孔,用平板震荡器振荡10min,让结晶物充分溶解,把空白对照组调零。用酶标仪以570nm/630nm双波长测定去除本底光吸收值后的吸光度(a)值,按下述公式计算出细胞增殖抑制率:
抑制率=(1-样品a值/对照组a值)×100%
所有实验重复3次后取平均值。结果如下述表3所示。
表3(单位为%):
实验例2:氧化荷苞牡丹碱对puc19质粒dna的作用
用tbe缓冲液(0.08mol/ltris-hcl,ph=8.5,0.08mol/l硼酸,0.0024mol/ledta)配制1%的琼脂糖凝胶溶液,然后加入一定量的gelred核酸染料;
对puc19质粒dna作用实验:加0.5μlpuc19质粒dna(0.5μg/μl)到灭菌处理过的微型塑料离心管中,再分别加不同浓度的化合物,用tris缓冲液定容至25μl,在恒温37℃下反应3h后,加入3μl溴酚蓝混合均匀,上样,以溴酚蓝溶液作为电泳时指示标记。在80v/cm电压下,电泳80分钟,最后在紫外灯下照射显影。
图2为化合物dco与dna作用的凝胶电泳图,由图2可知,随着化合物dco的浓度的逐渐增大,在浓度为100μm时,dna的超螺旋构型减少,dco与dna作用较强。随着化合物dco浓度的增大,dna的formⅰ泳带相对滞后,表明化合物dco嵌入到dna螺旋碱基对之间,并与之发生插入作用,使dna的相对分子量增大,导致泳带迁移率降低。与纯dna泳带(泳道1)相比,化合物体系(泳道2-5)的迁移率有所降低,发生滞后现象。这是化合物与dna发生插入作用的辅助证明。
- 还没有人留言评论。精彩留言会获得点赞!