一种间位酰氧基取代的苯乙酸类化合物及其合成方法和应用与流程
本发明公开涉及有机合成化学的技术领域,尤其涉及一种间位酰氧基取代的苯乙酸类化合物及其合成方法和应用。
背景技术:
过渡金属催化的碳氢键活化反应是实现碳-碳键及碳-杂原子键构建的一类重要手段,因其无需将底物预先官能化,具有原子经济型和步骤简洁等特点,在有机合成、药物合成及天然产物合成等方面得到了广泛的应用。近年来,研究人员主要通过向芳香化合物中引入含杂原子的导向基团,利用第二第三过渡金属与杂原子配位形成环金属络合物,以高选择性和高反应活性地实现芳香化合物特定位置的碳氢键活化反应。其中,大多数使用过渡金属钯、铑、镍、钴等为催化剂,对导向基的邻位进行碳氢键反应,实现碳-碳键及碳-杂原子键的构建。并且通过设计不同的导向基团,利用导向基团参与到反应中合成一系列稠杂环化合物,在一定的条件下,可以实现导向基团的脱除,进而不会影响底物的活性。
目前为止,对导向基邻位碳氢键活化反应有着较多的报道及较为深刻的研究,但对导向基团的间位、对位的碳氢键官能化反应的报道较少,主要是间位碳氢键活化反应过程中形成的大环金属过渡态能量较高,不是很稳定等因数导致反应较难实现。而利用过渡金属催化实现苯乙酸衍生物的乙酰氧基化反应更是未见报道。
因此,如何研发一种利用过渡金属催化实现苯乙酸衍生物的乙酰氧基化反应,以弥补技术空白,成为人们亟待解决的问题。
技术实现要素:
鉴于此,本发明公开了一种间位酰氧基取代的苯乙酸类化合物及其合成方法和应用,以弥补苯乙酸衍生物的乙酰氧基化反应的技术空白。
本发明一方面提供了一种苯乙酸衍生物的乙酰氧基化反应的方法,以式(ⅰ)所示的苯乙酸衍生物为原料,以醋酸碘苯为氧化剂,在催化剂、配体、添加剂以及溶剂组成的混合液中反应,反应结束后,用旋转蒸发仪抽掉溶剂,获得粗产物;
将所述粗产物进行柱层析,获得产物,其中,所述柱层析过程中所用的洗脱剂为石油醚和乙酸乙酯的混合溶剂;
其中,r为烷基、烷氧基、三氟甲基、氰基、硝基、f、cl、br、i中的一种、两种或三种;
r’为氢、c1-c5烷基以及芳基中的一种或两种。
上述苯乙酸衍生物的乙酰氧基化反应的具体过程为:将反应器抽真空,通入氮气置换,在所述反应器中依次加入苯乙酸衍生物、醋酸碘苯、催化剂、配体、添加剂和溶剂后,加热反应12-24h,反应结束后,用旋转蒸发仪抽掉溶剂,获得粗产物;
将所述粗产物进行柱层析,获得产物,其中,所述柱层析过程中所用的洗脱剂为石油醚和乙酸乙酯的混合溶剂。
优选,所述催化剂为过渡金属;优选为醋酸钯、二氯(五甲基环戊二烯基)氯化铑(iii)二聚体、二氯(五甲基环戊二烯基)氯化铱(iii)二聚体或二氯双(4-甲基异丙基苯基)钌(ii)。
进一步优选,所述配体为n-乙酰甘氨酸、n-乙酰丙氨酸、n-乙酰缬氨酸、n-乙酰苯丙氨酸、n-乙酰亮氨酸中的一种或两种。
进一步优选,所述添加剂为醋酸、乙酸酐中的一种或两种。
进一步优选,所述溶剂为n,n-二甲基甲酰胺、二甲基亚砜、二氯甲烷、乙腈、1,4-二氧六环、1,2-二氯乙烷、四氢呋喃、乙醇、甲醇、三氟乙醇以及六氟异丙醇中的一种或两种。
进一步优选,所述反应的反应温度为20-140℃。
进一步优选,所述苯乙酸衍生物与醋酸碘苯的反应摩尔比为1:4。
通过上述方法获得的产物为间位酰氧基取代的苯乙酸类化合物,其结构通式如下:
其中,r为烷基、烷氧基、三氟甲基、氰基、硝基、f、cl、br、i中的一种、两种或三种;
r’为氢、c1-c5烷基以及芳基中的一种或两种。
该间位酰氧基取代的苯乙酸类化合物可用于止疼、抗炎以及退热药物的合成。
本发明提供的苯乙酸衍生物的乙酰氧基化反应的方法,利用u-型模板策略,以嘧啶作为新型的导向基模板,在催化剂条件下,实现了苯乙酸衍生物间位的直接乙酰氧基化反应,大大简化反应过程,降低反应成本,具有反应活性高、选择性好、底物适用范围广等优点。
应当理解的是,以上的一般描述和后文的细节描述仅是示例性和解释性的,并不能限制本公开。
附图说明
此处的附图被并入说明书中并构成本说明书的一部分,示出了符合本发明的实施例,并与说明书一起用于解释本发明的原理。
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,对于本领域普通技术人员而言,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
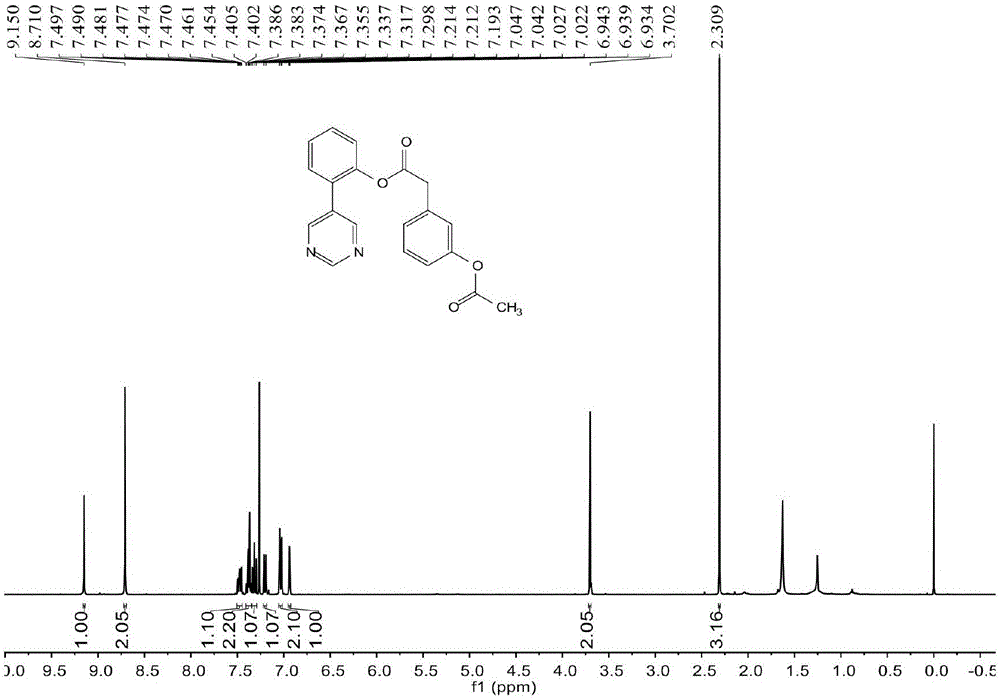
图1为本发明公开实施例1中化合物3a的氢谱图;
图2为本发明公开实施例1中化合物3a的碳谱图;
图3为本发明公开实施例2中化合物3b的氢谱图;
图4为本发明公开实施例2中化合物3b的碳谱图;
图5为本发明公开实施例3中化合物3c的氢谱图;
图6为本发明公开实施例3中化合物3c的碳谱图;
图7为本发明公开实施例4中化合物3d的氢谱图;
图8为本发明公开实施例4中化合物3d的碳谱图。
具体实施方式
下面以具体的实施方案对本发明进行进一步的解释,但是并不用于限制本发明的保护范围。
鉴于现有技术中,暂没有关于苯乙酸衍生物的乙酰氧基化反应的报道,本实施方案提供了一种苯乙酸衍生物的乙酰氧基化反应的方法,该方法利用u-型模板策略,以嘧啶作为新型的导向基模板,在催化剂条件下,实现了苯乙酸衍生物间位的直接乙酰氧基化反应,大大简化反应过程,具体而言,以式(ⅰ)所示的苯乙酸衍生物为原料,以醋酸碘苯为氧化剂,在催化剂、配体、添加剂以及溶剂组成的混合液中反应,反应结束后,用旋转蒸发仪抽掉溶剂,获得粗产物;
将所述粗产物进行柱层析,获得产物,其中,所述柱层析过程中所用的洗脱剂为石油醚和乙酸乙酯的混合溶剂;
其中,r为烷基、烷氧基、三氟甲基、氰基、硝基、f、cl、br、i中的一种、两种或三种,具体包括:间烷基、间烷氧基、间三氟甲基、间氰基、间硝基、间f、间cl、间br、间i、对烷基、对烷氧基、对三氟甲基、对氰基、对硝基、对f、对cl、对br以及对i;
r’为氢、c1-c5烷基以及芳基中的一种或两种。
上述苯乙酸衍生物的乙酰氧基化反应的具体反应操作过程如下:将反应器抽真空,通入氮气置换,在所述反应器中依次加入苯乙酸衍生物、醋酸碘苯、催化剂、配体、添加剂和溶剂后,加热反应12-24h,反应结束后,用旋转蒸发仪抽掉溶剂,获得粗产物;
将所述粗产物进行柱层析,获得产物,其中,所述柱层析过程中所用的洗脱剂为石油醚和乙酸乙酯的混合溶剂。
其中,催化剂为过渡金属,优选为:醋酸钯、二氯(五甲基环戊二烯基)氯化铑(iii)二聚体、二氯(五甲基环戊二烯基)氯化铱(iii)二聚体或二氯双(4-甲基异丙基苯基)钌(ii),该催化剂的用量为苯乙酸衍生物摩尔数的10%。
配体为n-乙酰甘氨酸、n-乙酰丙氨酸、n-乙酰缬氨酸、n-乙酰苯丙氨酸、n-乙酰亮氨酸中的一种或两种。
添加剂为醋酸、乙酸酐中的一种或两种,其中,用量为苯乙酸衍生物摩尔数的400%。
溶剂为n,n-二甲基甲酰胺、二甲基亚砜、二氯甲烷、乙腈、1,4-二氧六环、1,2-二氯乙烷、四氢呋喃、乙醇、甲醇、三氟乙醇以及六氟异丙醇中的一种或两种,其中,每毫摩尔反应物苯乙酸衍生物用溶剂5-20毫升。
上述间位乙酰氧基化方法中,加热反应的反应温度为20-140℃。
上述间位乙酰氧基化方法中,苯乙酸衍生物与醋酸碘苯的反应摩尔比为1:4。
其中,式(ⅰ)所示的苯乙酸衍生物可采用任何方法制备获得,只要满足其结构通式要求即可,以下为本发明所采用的制备方法,具体反应时如下:
具体的制备方法如下:步骤一:反应器抽真空通入氮气置换,依次加入5-溴嘧啶、2-羟基苯硼酸、醋酸钯、七水磷酸钾和异丙醇;80℃搅拌10-12h后,向反应器中加入盐水猝灭反应,所得产物用乙酸乙酯萃取并减压蒸馏,得到的粗产物经硅胶柱层析得到2-(5-嘧啶基)苯酚。步骤二:向反应容器中加入2-(5-嘧啶基)苯酚、芳基酰氯、三乙胺、dce;常温搅拌,所得产物用乙酸乙酯萃取并减压蒸馏,得到的粗产物经硅胶柱层析得到苯乙酸衍生物式(ⅰ)。
通过上述方法获得的产物为间位酰氧基取代的苯乙酸类化合物,其结构通式如下:
其中,r为烷基、烷氧基、三氟甲基、氰基、硝基、f、cl、br、i中的一种、两种或三种;
r’为氢、c1-c5烷基以及芳基中的一种或两种。
该间位酰氧基取代的苯乙酸类化合物可用于止疼、抗炎以及退热药物的合成。
在实验过程中,发明人将嘧啶基模板组装在药物分子布洛芬上,同样可以实现布洛芬衍生物的间位乙酰氧基化反应,嘧啶类导向基的模板可以在温和条件下移除,为药物分子的官能团化提供了新的合成路线,并且具有较高的间位选择性。
下面以具体的实施例对本发明进行更进一步的解释说明。
实施例1
反应器抽真空通入氮气置换三次,依次加入0.2mmol(58.0mg)苯乙酸衍生物(1a),0.8mmol(257.58mg)醋酸碘苯(2),0.02mmol(4.48mg)pd(oac)2,0.05mmol(5.86mg)ac-gly-oh,0.8mmol(81.7mg)乙酸酐,1mlhfip,100℃搅拌24h。反应结束后,用旋转蒸发仪抽掉溶剂,粗产物通过柱层析,洗脱剂为石油醚:乙酸乙酯=3:1混合溶剂,得到35.5mg间乙酰氧基取代的苯乙酸衍生物3a,分离收率为51%。
化合物3a的表征数据如下:
1hnmr(400mhz,cdcl3)δ9.15(s,1h),8.71(s,2h),7.50-7.45(m,1h),7.41-7.36(m,2h),7.32(t,j=8.0hz,1h),7.21-7.19(m,1h),7.03(dd,j=7.9,2.0hz,2h),6.94(t,j=2.0hz,1h),3.70(s,2h),2.31(s,3h).13cnmr(100mhz,cdcl3)δ169.4,169.0,157.6,156.3,150.9,147.9,134.0,131.1,130.5,130.4,129.8,127.9,127.0,126.6,123.2,122.4,120.8,40.9,21.2.hrms(esi):calcdforc20h16n2o4([m+h]+)349.1183,found349.1192.(参见图1、图2)。
实施例2
反应器抽真空通入氮气置换三次,依次加入0.2mmol(64.02mg)苯乙酸衍生物(1b),0.8mmol(257.58mg)醋酸碘苯(2),0.02mmol(4.48mg)pd(oac)2,0.05mmol(5.86mg)ac-gly-oh,0.8mmol(81.7mg)乙酸酐,1mlhfip,100℃搅拌24h。反应结束后,用旋转蒸发仪抽掉溶剂,粗产物通过柱层析,洗脱剂为石油醚:乙酸乙酯=3:1混合溶剂,得到41.6mg间乙酰氧基取代的苯乙酸衍生物3b,分离收率为55%。
化合物3b的表征数据如下:
1hnmr(400mhz,cdcl3)δ9.18(s,1h),8.75(s,2h),7.49-7.45(m,1h),7.38-7.36(m,2h),7.23(d,j=8.0hz,1h),7.00(dd,j=8.8,2.8hz,1h),6.88(d,j=2.8hz,1h),6.83(d,j=8.9hz,1h),3.78(s,3h),3.70(s,2h),2.28(s,3h).13cnmr(100mhz,cdcl3)δ169.9,169.3,157.4,156.4,155.1,148.0,143.8,131.3,130.5,130.3,127.7,126.8,124.0,123.3,122.6,121.6,110.9,55.8,35.9,21.1.hrms(esi):calcdforc21h18n2o5([m+h]+)379.1288,found379.1297.(见图3、图4)。
实施例3
反应器抽真空通入氮气置换三次,依次加入0.2mmol(60.8mg)苯乙酸衍生物(1c),0.8mmol(257.58mg)醋酸碘苯(2),0.02mmol(4.48mg)pd(oac)2,0.05mmol(5.86mg)ac-gly-oh,0.8mmol(81.7mg)乙酸酐,1mlhfip,100℃搅拌24h。反应结束后,用旋转蒸发仪抽掉溶剂,粗产物通过柱层析,洗脱剂为石油醚:乙酸乙酯=3:1混合溶剂,得到39.1mg间乙酰氧基取代的苯乙酸衍生物3c,分离收率为54%。
化合物3c的表征数据如下:
1hnmr(400mhz,cdcl3)δ9.12(s,1h),8.70(s,2h),7.50-7.46(m,1h),7.40-7.33(m,2h),7.21(dd,j=8.1,0.8hz,1h),6.83(s,2h),6.72(s,1h),3.65(s,2h),2.33(s,3h),2.29(s,3h).13cnmr(100mhz,cdcl3)δ169.8,169.1,157.2,156.3,150.7,147.8,140.2,133.6,131.2,130.5,130.4,127.7,127.5,127.0,123.2,121.5,119.4,40.9,21.3,21.1.hrms(esi):calcdforc21h18n2o4([m+h]+)363.1339,found363.1348.(见图5、图6)。
实施例4:
反应器抽真空通入氮气置换三次,依次加入0.2mmol(64.02mg)苯乙酸衍生物(1d,0.8mmol(257.58mg)醋酸碘苯(2),0.02mmol(4.48mg)pd(oac)2,0.05mmol(5.86mg)ac-gly-oh,0.8mmol(81.7mg)乙酸酐,1mlhfip,100℃搅拌24h。反应结束后,用旋转蒸发仪抽掉溶剂,粗产物通过柱层析,洗脱剂为石油醚:乙酸乙酯=3:1混合溶剂,得到36.3mg间乙酰氧基取代的苯乙酸衍生物3d,分离收率为48%。
化合物3d的表征数据如下:
1hnmr(400mhz,cdcl3)δ9.15(s,1h),8.71(s,2h),7.48-7.45(m,1h),7.40-7.35(m,2h),7.22-7.19(m,1h),6.59(t,j=2.0hz,1h),6.57(t,j=2.0hz,1h),6.54(t,j=1.6hz,1h),3.78(s,3h),3.65(s,2h),2.30(s,3h).13cnmr(100mhz,cdcl3)δ169.3,168.9,160.6,157.6,156.3,151.7,147.9,134.6,131.1,130.5,130.4,127.9,127.0,123.2,114.8,112.5,106.9,55.5,41.1,21.2.hrms(esi):calcdforc21h18n2o5([m+h]+)379.1288,found379.1295.(见图7、图8)。
本领域技术人员在考虑说明书及实践这里公开的发明后,将容易想到本发明的其它实施方案。本申请旨在涵盖本发明的任何变型、用途或者适应性变化,这些变型、用途或者适应性变化遵循本发明的一般性原理并包括本发明未公开的本技术领域中的公知常识或惯用技术手段。说明书和实施例仅被视为示例性的,本发明的真正范围和精神由权利要求指出。
应当理解的是,本发明并不局限于上面已经描述的内容,可以在不脱离其范围进行各种修改和改变。本发明的范围仅由所附的权利要求来限制。
- 还没有人留言评论。精彩留言会获得点赞!
- 2,3,4‑三乙酰基‑1‑(5‑溴‑2‑吡啶基)巯基‑α‑L‑岩藻吡喃糖苷的制造方法与工艺
- 一种他洛柳酯药物中间体2‑乙酰氧基苯甲酸‑3‑羟基苯酞酯的合成方法与制造工艺
- 一种生产2‑(N‑甲基氯乙酰氨基)‑5‑氯二苯甲酮的方法与制造工艺
- 由三乙酰5‑甲基尿苷合成2′,3′‑二脱氢‑3′‑脱氧胸苷的方法与制造工艺
- 合成1β‑甲基碳青霉烯类抗生素双环母核的关键中间体4‑BMA的制备方法与制造工艺
- γ晶型利福昔明的制备方法和用途与制造工艺
- 一种盐酸普拉格雷化合物及其制备方法和含有盐酸普拉格雷的药物组合物与制造工艺
- 包含可氢化材料和聚合基质的氢储器的制造方法与工艺
- 一种药物中间体的合成方法与制造工艺
- 一种硅烷偶联剂的水解装置的制造方法