一种抗丙肝药物雷迪帕韦关键中间体的制备方法与流程
本发明涉及一种抗丙肝药物雷迪帕韦关键中间体的制备方法,即1-(7-溴-9,9-二氟-9h-芴-2-基)-2-氯乙酮的制备方法。
背景技术:
雷迪帕韦(ledipasvir),化学名:n-[(2s)-1-[(6s)-6-[5-[9,9-二氟-7-[2-[(1s,2s,4r)-3-[(2s)-2-(甲氧基羰基氨基)-3-甲基丁酰基]-3-氮杂双环[2.2.1]-2-庚基]-3h-苯并咪唑-5基]-2-芴基]-1h-2-咪唑基]-5-氮杂螺烷[2.4]-5-庚基]-3-甲基-1-氧代-2-丁基]氨基甲酸甲酯,是由吉利德科学公司开发,与该公司另一抗丙肝重磅产品sovaldi(通用名:索非布韦)复方组合,商品名:harvoni(复方索非布韦/雷迪帕韦),美国fda于2014年10月10日批准其上市,同年11月21日获欧盟上市许可,是一种全口服,一天一次的片剂,用于治疗基因1型的丙型肝炎感染。harvoni既可单药使用,也可以和其它口服制剂比如利巴韦林联合使用。
7-溴-2-(氯乙酰基)-9,9-二氟芴作为雷迪帕韦的关键中间体,其合成方案主要有以下三种方法:方案1以2-溴芴为起始原料,经碘代,氟代,与2-氯-n-甲氧基-n-甲基乙酰胺反应制得。该方案原料较贵,并且用到格氏试剂,存在危险工艺。如:u.s.pat.appl.publ.,20130324496。方案2同样以2-溴芴为起始原料,与氯乙酰氯经付克酰基化,羰基保护,氟代,脱保护制得。该方案同样原料较贵。如:pctint.appl.,2016078505。方案3同样以2-溴芴为起始原料,与乙酰氯经付-克酰基化,羰基保护,氟代,脱保护,氯代制得。该方案同样原料较贵,反应路线较长,收率不高。如:pctint.appl.,2016078505。具体路线如下:
方案1:
方案2:
方案3:
技术实现要素:
本发明的主要目的是:提供一种成本低廉,操作简便的雷迪帕韦关键中间体1-(7-溴-9,9-二氟-9h-芴-2-基)-2-氯乙酮的制备方法。
本发明的合成路线如下:
技术方案:一种雷迪帕韦关键中间体1-(7-溴-9,9-二氟-9h-芴-2-基)-2-氯乙酮的制备方法,包括如下步骤:
第一步:2-碘-5-溴苯甲酸的合成(2):
2-氨基-5-溴苯甲酸经重氮化,合成重氮盐,然后与碘化钠反应合成2-碘-5-溴苯甲酸(2);具体反应如下:
第二步:2-碘-5-溴苯甲酸甲酯的合成(3):
2-碘-5-溴苯甲酸首先与氯化亚砜反应合成酰氯,然后与甲醇反应形成酯2-碘-5-溴苯甲酸甲酯(3);具体反应如下:
第三步:5-溴-2-苯基苯甲酸甲酯的合成(4):
2-碘-5-溴苯甲酸甲酯与苯硼酸经suzuki偶联合成5-溴-2-苯基苯甲酸甲酯(4);具体反应如下:
第四步:5-溴-2-苯基苯甲酸的合成(5):
5-溴-2-苯基苯甲酸甲酯经碱水解制得5-溴-2-苯基苯甲酸(5);具体反应如下:
第五步:2-溴-9-芴酮的合成(6):
5-溴-2-苯基苯甲酸首先与氯化亚砜反应合成酰氯,然后经分子内付克酰基化反应制得2-溴-9-芴酮(6);具体反应如下:
第六步:2-溴芴的合成(7):
2-溴-9-芴酮在冰乙酸中,经pd/c催化氢化制得2-溴芴(7);具体反应如下:
第七步:2-溴-7-碘芴的合成(8):
2-溴芴与碘酸钾与碘在冰乙酸溶液中制备2-溴-7-碘芴(8);具体反应如下:
第八步:2-溴-7-碘-9,9-二氟芴的合成(9):
2-溴-7-碘芴与n-氟代双苯磺酰胺反应制备2-溴-7-碘-9,9-二氟芴(9);具体反应如下:
第九步:1-(7-溴-9,9-二氟-9h-芴-2-基)-2-氯乙酮的合成(1)
2-溴-7-碘-9,9-二氟芴与2-氯-n-甲氧基-n-甲基乙酰胺反应制得1-(7-溴-9,9-二氟-9h-芴-2-基)-2-氯乙酮(1);具体反应如下:
作为优化:所述的第一步:2-碘-5-溴苯甲酸的合成(2)中,投料量:2-氨基-5-溴苯甲酸:nano2:naoh:ki=1:1.1-1.2:1.05-1.1:1.4-1.5;重氮化反应温度0-5℃,反应时间2-3h;碘代反应温度,35-40℃加入ki硫酸溶液,90℃反应1-2h;重结晶条件:30%-70%乙醇溶液。
作为优化:所述的第二步:2-碘-5-溴苯甲酸甲酯的合成(3)中,投料量:2-碘-5-溴苯甲酸:氯化亚砜:甲醇=1:1.3-2:3-5;反应温度室温,反应时间3-5h。
作为优化:所述的第三步:5-溴-2-苯基苯甲酸甲酯的合成(4)中,投料量:pd(pph3)2cl2:苯硼酸:碳酸钠:4-溴-2-碘苯甲酸甲酯=0.01:1.05-1.1:2-3:1;溶剂:thf/水;反应温度室温至80℃。
作为优化:所述的第四步:5-溴-2-苯基苯甲酸的合成(5)中,投料量:4-溴-2-甲酸甲酯基-1,1-联苯:氢氧化钠=1:1.8-2;溶剂:甲醇-水;反应温度:60-80℃,反应时间4-8h。
作为优化:所述的第五步:2-溴-9-芴酮的合成(6)中,投料量:5-溴-2-苯基苯甲酸:氯化亚砜:无水三氯化铝=1:1.5-2:1.05-1.1,反应温度:制备酰氯温度室温,付克反应温度0℃-室温。
作为优化:所述的第六步:2-溴芴的合成(7)中,投料量:2-溴-9-芴酮:pd/c=1:0.01;反应温度为60℃,反应时间:5-7h。
作为优化:所述的第七步:2-溴-7-碘芴的合成(8)投料量:2-溴芴:碘(1.1g,4.5mmol):碘酸钾=1:0.2-0.25:0.3-0.4;溶剂:冰乙酸:硫酸;反应温度80℃,反应时间10-12h。
作为优化:所述的第八步:2-溴-7-碘-9,9-二氟芴的合成(9)中,投料量:2-溴-7-碘芴:n-氟代双苯磺酰胺:双-(三甲基硅基)胺锂=1:2.3-2.7:3-3.1。
作为优化:所述的第九步:1-(7-溴-9,9-二氟-9h-芴-2-基)-2-氯乙酮的合成(1)中,投料量:中间体13:异丙基氯化镁:2-氯-n-甲氧基-n-甲基乙酰胺=1:1.1-1.3:1.1-1.2;重结晶条件:70%-90%甲醇溶液。
有益效果:本发明以2-氨基-5-溴苯甲酸为原料,经重氮化、碘代、合成5-溴-2-碘苯甲酸、甲酯化、与苯硼酸偶联反应、酯水解、酰氯化、分子内付-克酰基化、羰基还原、碘代、氟代、最后与2-氯-n-甲氧基-n-甲基乙酰胺反应制得目标产物。本工艺起始原料易得,价格低廉,无危险工艺,反应条件温和。
附图说明
图1是本发明的结构示意图。
具体实施方式
下面将对本发明实施例中的技术方案进行清楚、完整地描述,以使本领域的技术人员能够更好的理解本发明的优点和特征,从而对本发明的保护范围做出更为清楚的界定。本发明所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例,基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动的前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例1、2-碘-5-溴苯甲酸的合成(2)
取2-氨基-5-溴苯甲酸(2.14g,10mmol),nano2(0.828g,12mmol)和naoh(0.55g,11mmol)溶于40ml水中,搅拌下,冰浴冷却至0℃,滴加12ml6mol/l的盐酸溶液,2h滴完。盐酸滴加完毕后,反应体系在0℃下继续反应1h,然后反应体系升温至35-40℃,缓慢加入ki的溶液[(ki2.5g,15mmol),h2so4(0.6ml)和水(5ml)],20min加完。然后反应体系升温至90℃反应1h。反应结束后,反应体系缓慢冷却至室温,析出大量固体,过滤,水洗,得黄色粗品。粗品经50%乙醇溶液重结晶得浅黄色固体2.87g,收率87%。m.p.158-159℃。
1hnmr(400mhz,cdcl3):δ8.15(d,j=2.5hz,1h),7.90(d,j=8.5hz,1h),7.34(dd,j=2.5,8.5hz,1h);13cnmr(100mhz,cdcl3):δ166.9,142.3,139.1,135.1,132.3,121.4,92.9。
实施例2、2-碘-5-溴苯甲酸甲酯的合成(3)
取2-碘-5-溴苯甲酸(3.25g,10mmol),溶于30ml二氯甲烷中,加入氯化亚砜(1.8g,15mmol),dmf3滴,室温搅拌3h,反应结束后,减压除去溶剂和过量的氯化亚砜,残留物加入30ml二氯甲烷溶解,冰浴下缓慢加入20ml甲醇,加完后,反应体系升温至室温继续搅拌反应1h,反应结束后,冰浴下,加入饱和的碳酸钠水溶液40ml,搅拌5min,分液,滤液水洗至中性,有机层无水硫酸镁干燥,过滤,滤液减压浓缩,残留物柱层析得浅黄色固体3.32g,收率98%。m.p.45-47℃。
1hnmr(400mhz,cdcl3):δ7.95(s,1h),7.86(d,j=8hz,1h),7.31(d,j=8hz,1h),3.96(s,3h);13cnmr(100mhz,cdcl3):δ165.6,142.7,136.5,135.7,133.9,122.3,92.19,52.8。
实施例3、5-溴-2-苯基苯甲酸甲酯的合成(4)
取pd(pph3)2cl2(70mg,0.1mmol),苯硼酸(1.28g,10.5mmol),碳酸钠(2.12g,20mmol),水30ml和thf30ml。氩气保护下,反应体系在室温条件下搅拌5min,然后加入4-溴-2-碘苯甲酸甲酯(3.39g,10mmol)。然后反应体系升温至80℃反应8h。反应结束后,反应体系缓慢冷却至室温,二氯甲烷萃取(50ml*3),合并有机相,水洗,有机层无水硫酸钠干燥,过滤,滤液减压回收溶剂,残留物柱层析得无色液体2.44g,收率86%。
1hnmr(400mhz,cdcl3):δ7.99(d,j=2.0hz,1h),7.67(dd,j=8.4,2.0hz,1h),7.44–7.38(m,3h),7.31–7.28(m,3h),3.67(s,3h);13cnmr(100mhz,cdcl3):δ167.6,141.4,140.1,134.2,132.6,132.4,132.3,128.2,127.6,121.01,52.2。
实施例4、5-溴-2-苯基苯甲酸的合成(5)
取4-溴-2-甲酸甲酯基-1,1-联苯(2.9g,10mmol),溶于20ml甲醇,加入氢氧化钠(0.8g,20mmol),水20ml,80℃反应4h,反应结束后,6mol/l的盐酸调节体系ph至2-3,过滤,滤饼水洗至中性,干燥得白色固体2.7g,收率98%。m.p.151-152℃。
1hnmr(400mhz,cdcl3):δ8.09(d,j=2.1hz,1h),7.69(dd,j=8.2,2.1hz,1h),7.41–7.37(m,3h),7.31–7.29(m,2h),7.25(d,j=8.2hz,1h).13cnmr(100mhz,cdcl3):δ172.3,142.28,139.8,135.1,133.4,132.8,130.7,128.3,128.2,127.7,121.1。
实施例5、2-溴-9-芴酮的合成(6)
取5-溴-2-苯基苯甲酸(2.76g,10mmol),二氯甲烷30ml,氯化亚砜(2.36g,20mmol)和加入3滴dmf,室温反应8h,反应结束后,减压回收溶剂及除去未反应完的氯化亚砜,残留物加入50ml乙酸乙酯,冰浴下,分批加入无水三氯化铝(1.5g,12mmol),反应体系缓慢升温至室温继续反应6h,反应结束后,反应物缓慢倒入冰水中,加入浓盐酸调制ph至2-3,分液,有机层水洗至中性,过滤,滤液减压回收溶剂,残留物柱层析得黄色固体2.35g,收率91%,145-146℃。
1hnmr(400mhz,cdcl3):δ7.63(s,1h),7.59(d,j=7.2hz,1h),7.49(d,j=7.2hz,1h),7.41(t,j=7.2hz,1h),7.36(d,j=6.8hz,1h),7.24(t,j=6.0hz,2h);13cnmr(100mhz,cdcl3):δ192.0,143.3,142.7,136.8,135.5,134.8,133.4,129.2,127.2,124.3,122.7,121.5,120.2。
实施例6、2-溴芴的合成(7)
取2-溴-9-芴酮(2.58g,10mmol),溶于30ml冰乙酸中,加入pd/c0.1g,通入h2,升温至60℃反应7h。反应结束后,过滤,滤液减压浓缩,残留物水洗至中性,残留物柱层析得白色固体2.26g,收率93%.m.p.110-111℃。
1hnmr(400mhz,cdcl3):δ7.78-7.58(m,3h),7.56-7.46(m,2h),7.42–7.28(m,2h),3.83(s,2h).13cnmr(100mhz,cdcl3):δ145.4,143.0,140.9,130.0,128.4,127.3,127.1,125.2,121.3,120.7,120.1,36.9。
实施例7、2-溴-7-碘芴的合成(8)
取2-溴芴(2.44g,10mmol),碘(1.1g,4.5mmol)以及碘酸钾(0.65g,3mmol)溶于(冰乙酸45ml,水5ml和硫酸2ml)的混合溶剂中。氮气保护下,反应体系升温至80℃反应12h,反应结束后,反应体系缓慢降温至室温,10ml二氯甲烷萃取,水洗至中性。有机层无水硫酸钠干燥,过滤,滤液减压回收溶剂,残留物柱层析得白色固体3.63g,收率98%。
1hnmr(400mhz,cdcl3):δ7.86(s,1h),7.70-7.65(m,2h),7.59(d,j=8.0hz,1h),7.51-7.47(m,2h),3.83(s,2h);13cnmr(100mhz,cdcl3):δ145.3,144.9,140.6,140.1,136.3,134.5,130.5,128.6,121.9,121.6,92.6,36.8。
实施例8、2-溴-7-碘-9,9-二氟芴的合成(9)
取2-溴-7-碘芴(3.7g,10mmol),n-氟代双苯磺酰胺(nfsi)(7.9g,25mmol)。氮气保护下,加入thf100ml,反应液冷却至-78℃,滴加双-(三甲基硅基)胺锂的thf溶液(4.9g,30mmol),滴加过程中保持温度在-60℃以下,滴加完毕后,继续反应3h,反应结束后,反应液用nh3/ch3oh淬灭,移除低温浴槽,反应体系中有混浊物出现,过滤,thf洗涤,收集滤液,减压蒸干,残留物加入二氯甲烷回流搅拌,过滤,滤液减压回收溶剂,残留物加入80%甲醇水溶剂打浆,过滤得黄色固体3.3g,收率81%.m.p.139-141℃。
1hnmr(400mhz,cdcl3):δ7.94-7.74(m,3h),7.60-7.29(m,3h).13cnmr(100mhz,cdcl3):δ149.7,146.8,141.4,141.1,137.5,134.8,131.3,129.4,124.3,122.5,121.9,93.8。
实施例9、1-(7-溴-9,9-二氟-9h-芴-2-基)-2-氯乙酮的合成(1)
氮气保护下,取中间体1340.5g,溶于200ml无水thf中,反应体系冷却至-15°,滴加13g异丙基氯化镁的thf溶液80ml,滴加过程中,控制反应温度在-5°以下,滴加完毕后,继续反应1h,之后滴加入15g2-氯-n-甲氧基-n-甲基乙酰胺的thf溶液60ml,滴加过程中控制反应温度不超过-5°,滴加结束后,将体系温度升至-5-0°反应,反应结束后,0°条件下加入2n盐酸淬灭反应,加入乙酸乙酯200ml,,分液,有机层水洗,无水硫酸钠干燥,过滤,减压回收溶剂,残留物,用甲醇:水=10:1重结晶得白色固体27g,收率76%。
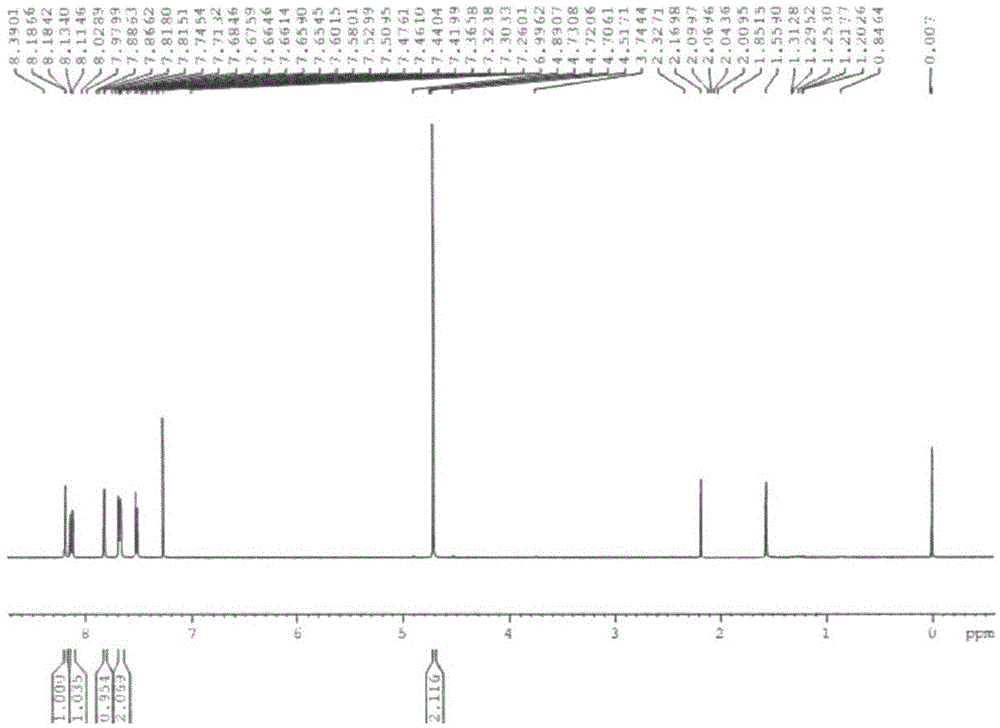
1hnmr(400mhz,cdcl3):δ8.2-8.13(m,2h),7.84(s,1h),7.67(m,2h),7.53(m,1h),4.72(s,2h)。
- 还没有人留言评论。精彩留言会获得点赞!