具有抗肿瘤活性的2-甲氧基烟酰胺衍生物制备方法及用途与流程
本发明属于抗肿瘤新化合物合成及药物应用
技术领域:
,具体涉及一种具有抗肿瘤活性的2-甲氧基烟酰胺衍生物制备方法及用途。
背景技术:
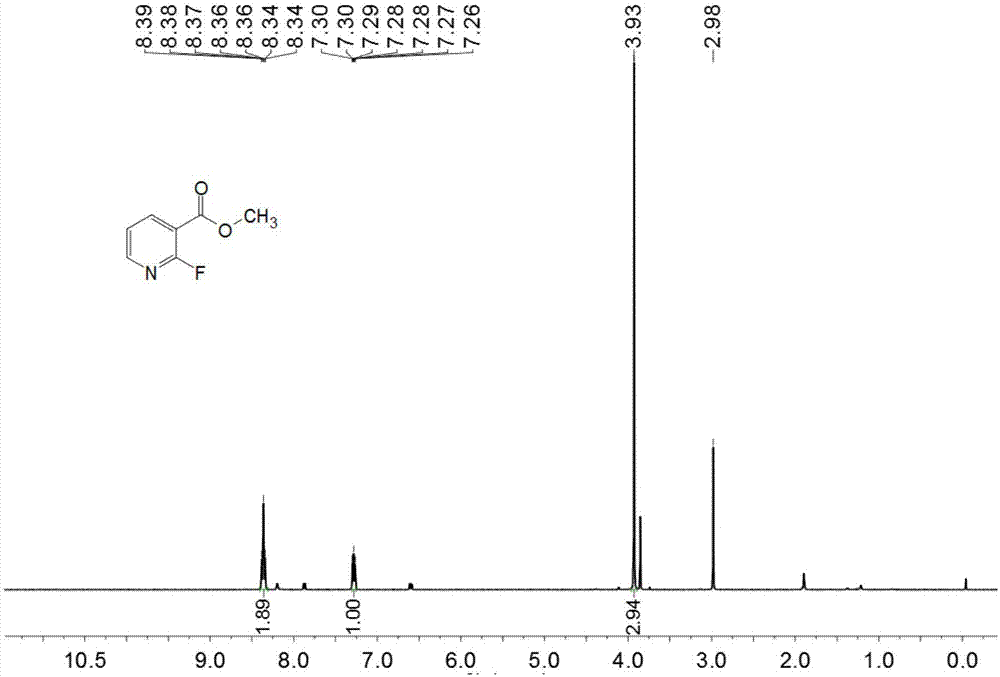
:随肝癌是消化系统常见的恶性肿瘤之一,占恶性肿瘤死亡率第3位。在过去的几十年里,全世界肝癌的发病率正逐年增加,每年新增病例近75万。中国是肝癌高发病地区,在近年的全国肿瘤登记中,我国肝癌的死亡率高居第2位。肝癌俨然已成为严重危害全世界人类健康的流行性疾病,给各国的医疗保障、家庭和个人造成了极大的经济和精神负担。目前肝癌临床化学治疗主要有两种途径:细胞毒性药物和分子靶向药物。细胞毒性药物抑制肿瘤细胞生长的同时,也严重抑制了正常细胞的增殖,因此毒副作用较大;针对特定底物蛋白的靶向药物能够有效地抑制肿瘤细胞增殖,具有特异性高、选择性强和非细胞毒性等优点。因此,近年来随着对肝癌发生、发展、扩散机制中多种细胞信号途径的深入研究,利用分子靶向治疗,即利用具有特异性靶点的单克隆抗体或激酶抑制剂靶向于肝癌信号通路的关键分子,特异性地阻断信号转导,从而抑制肝癌细胞的生长与增殖,发挥抗癌作用,这一治疗方法得到了迅速的发展。烟酰胺是一种水溶性维生素,也是辅酶ⅰ和辅酶ⅱ的组成部分,它可以参与体内脂质代谢,组织呼吸的氧化过程和糖类无氧分解的过程,在临床上被广泛的用于防治糙皮病、舌炎、皮炎等疾病。近年来,基于药理活性基础上的烟酰胺类衍生物的合成在医药及农药研究领域得到了快速发展,尤其是2-甲氧基烟酰胺衍生物在药物分子研究中得到了独特的关注。1,2,4-恶二唑作为一类结构特殊的杂环化合物,广泛存在于自然界中,其衍生物具有抑菌、抗炎、抗病毒、降压等多种生理活性,作为一种有效的药物活性基团,被广泛的应用于新药研究领域。cn200810063017.6公开了6-芳基-3-取代羧基吡啶类化合物的药物用途,其结构通式如式(ⅱ)所示,本通式化合物具有显著抑制体外培育人慢性髓原细胞白血病细胞株(k562)和小鼠淋巴样瘤细胞株(p388d1)生长的活性,可用于治疗白血病及其相关肿瘤性疾病。本发明通过将2-甲氧基烟酰胺与1,2,4-恶二唑片段链接起来构筑新的结构骨架,得到结构新颖的n-(2,6-二氯-4-(1,2,4–恶二唑-3-基)苯基)-2-甲氧基烟酰胺,其具有明显的抑制肿瘤细胞生长的活性,尤其是对人肝癌hepg2细胞具有优异的抑制活性。技术实现要素:本发明提供式(ⅰ)所示化合物。同时,本发明提供式(ⅰ)所示化合物的制备方法,其包含如下步骤:其中,催化剂为中强酸,选自三氟乙酸、甲磺酸、对甲苯磺酸、冰醋酸、多聚磷酸。式(ⅰ)所示化合物的制备方法还包括如下步骤:其中,缚酸剂为有机胺,选自三乙胺、二甲异丙胺、二异丙基乙基胺、吡啶、三乙烯二胺。式(ⅰ)所示化合物的制备方法还包括如下步骤:其中,强碱选自lihmds(六甲基二硅基氨基锂)、正丁基锂、仲丁基锂。式(ⅰ)所示化合物的制备方法还包括如下步骤:其中,无机碱选自碳酸钾、碳酸钠、氢氧化钠、氢氧化钾。本发明提供式(ⅰ)所示化合物在制备抗肿瘤药物中用途,尤其是抗肝癌、结肠癌、肺癌、乳腺癌、宫颈癌用途。本发明化合物对人肝癌细胞hepg2,结肠癌细胞lovo细胞,人非小细胞肺癌a549细胞,人乳腺癌mcf-7,sk-br-3细胞,人宫颈癌hela细胞均具有较明显的抑制活性,尤其是对人肝癌hepg2细胞具有优异的抑制活性,而且毒性较低,可作为抗肿瘤性疾病的药物。附图说明图1是式(3)所示化合物的1h-nmr图图2是式(5)所示化合物的1h-nmr图图3是式(6)所示化合物的1h-nmr图图4是式(ⅰ)所示化合物的1h-nmr图图5是式(ⅰ)所示化合物的13c-nmr图图6是式(ⅰ)所示化合物的单晶(xrd)结构图图7是式(ⅰ)所示化合物的治疗不同时间点肿瘤的体积变化图图8是式(ⅰ)所示化合物的两组治疗结束后肿瘤的重量图图9是式(ⅰ)所示化合物的治疗不同时间点实验小鼠的体重变化图图10是式(ⅰ)所示化合物的两组治疗结束后肿瘤的大小图具体实施方式以下实施方式旨在说明本发明,而不是对发明的进一步限定。实施例12-氟盐酸甲酯的合成在250ml的单口瓶中加入2-氟烟酸(7.05g,50mmol)和dmf(60ml),在室温条件下缓慢滴加硫酸二甲酯(7.56g,60mmol),滴加完毕后,升温至50℃反应8h。tlc监测反应结束后,加入饱和食盐水(100ml)稀释、乙酸乙酯(3×50ml)萃取、合并有机相、无水硫酸钠干燥,有机相经减压旋蒸后,粗品经硅胶柱层析分离(v石油醚:v乙酸乙酯=10:1)得到式(3)化合物,淡黄色油状液体5.83g,收率92.5%。1hnmr(400mhz,cdcl3)δ8.40~8.31(m,2h),7.31~7.25(m,1h),3.93(s,3h),具体见附图1。实施例2n-(2,6-二氯-4-氰基苯基)-2-甲氧基烟酰胺的合成在100ml的单口瓶中加入4-氨基-3,5-二氯苯腈(1.86g,10mmol)和二氧六环(40ml),在0℃条件下缓慢滴加14mllihmds/thf溶液(14mmol,1mol/l),搅拌均匀后,缓慢滴加式(3)化合物(1.38g,11mmol)的二氧六环溶液(10ml),滴加完毕后,升温至回流反应14h。tlc监测反应结束后,加入饱和氯化铵水溶液(10ml)淬灭反应,有机相经减压旋蒸后,粗品经硅胶柱层析分离(v石油醚:v乙酸乙酯=1:1)得到式(5)化合物1.82g,收率56.8%。1hnmr(400mhz,cdcl3)δ9.84(s,1h),8.60(dd,j=7.6,2.0hz,1h),8.41(dd,j=4.8,2.0hz,1h),7.74(s,2h),7.17(dd,j=7.6,4.9hz,1h),4.24(s,3h);13cnmr(100mhz,cdcl3)δ163.35,162.56,152.81,144.17,139.42,136.00,133.65,120.10,118.05,116.67,113.97,56.49,具体见附图2。实施例3(z)-n-[2,6-二氯-4-(n'-羟基脒)苯基]-2-甲氧基烟酰胺的合成在25ml的单口瓶中加入式(5)化合物(161mg,0.5mmol)、甲醇(10ml)、三乙胺(151mg,1.5mmol)和盐酸羟胺(104mg,1.5mmol)。反应体系在室温条件下搅拌2h,体系呈现白色浑浊,减压抽滤,并用冰的甲醇洗涤,真空干燥得到式(6)化合物116mg,收率65.6%。1hnmr(400mhz,dmso-d6)δ10.09(s,1h),10.01(s,1h),8.40(dd,j=4.9,1.9hz,1h),8.21(dd,j=7.4,1.9hz,1h),7.84(s,2h),7.19(dd,j=7.4,4.9hz,1h),6.07(s,2h),4.04(s,3h),具体见附图3。实施例4n-[2,6-二氯-4-(1,2,4-恶二唑-3-基)苯基]-2-甲氧基烟酰胺的合成在10ml的单口瓶中加入式(6)化合物(71mg,0.2mmol)、原甲酸三甲酯(3ml)和三氟乙酸(0.1ml)。反应体系在室温条件下搅拌30min,然后加热到60℃,继续反应4.5h。tlc监测反应结束后减压旋蒸,粗品经硅胶柱层析分离(v石油醚:v乙酸乙酯=1:1)得到式(ⅰ)化合物69mg,收率94.2%。1hnmr(400mhz,cdcl3)δ9.77(s,1h),8.83(s,1h),8.63(dd,j=7.5,1.6hz,1h),8.43~8.37(m,1h),8.22(s,2h),7.16(dd,j=7.5,4.9hz,1h),4.24(s,3h);13cnmr(100mhz,cdcl3)δ165.76,165.22,161.71,160.76,150.61,142.27,135.25,133.95,127.39,126.48,118.10,115.14,54.50,具体见附图4和附图5。实施例5n-[2,6-二氯-4-(1,2,4-噁二唑-3-基)苯基]-2-甲氧基烟酰胺的单晶加热条件下,取100mgn-[2,6-二氯-4-(1,2,4-噁二唑-3-基)苯基]-2-甲氧基烟酰胺(i)溶于50ml石油醚与乙酸乙酯的混合溶剂(v石油醚/v乙酸乙酯=1/1),室温放置自然挥发,7-10天后析出n-[2,6-二氯-4-(1,2,4-噁二唑-3-基)苯基]-2-甲氧基烟酰胺(i)晶体,得式(ⅰ)化合物单晶。实施例6n-[2,6-二氯-4-(1,2,4-噁二唑-3-基)苯基]-2-甲氧基烟酰胺的单晶测定选取0.36mm×0.2mm×0.1mm的单晶体,在brukerapx-iiccd型衍射仪上收集衍射数据,利用石墨单色器单色化的mo-kα射线在173(2)k下以φ-ω扫描方式收集衍射数据,在2.7≤θ≤27.4°范围内收集6788个数据,其中独立衍射点3366个,可观测点2780个,然后用bruker的saintv8.34a程序还原数据,用sadabs程序进行经验吸收校正。应用shelxs-97(sheldrick,2008)程序直接法解析和精修结构,所有的非氢原子采用全矩阵最小二乘法进行结构精修,所有非氢原子都做各向异性精修,理论加氢,氢原子各向同性热参数修正,具体情况见表1至表6。测定结果见附图6。表1式(ⅰ)化合物的晶体数据表2式(ⅰ)化合物的数据收集参数表3式(ⅰ)化合物的精修参数表4式(ⅰ)化合物晶体的原子坐标(×104)和热参数表5式(ⅰ)化合物晶体的部分键长键(bond)键长(dist.)键(bond)键长(dist.)cl1—c71.7325(19)n2—c21.381(2)cl2—c51.7332(19)n2—c11.296(3)o3—c141.354(2)c5—c41.386(2)o3—c151.447(2)o1—n11.409(2)o2—c91.220(2)o1—c11.328(3)n4—c141.324(2)c13—c121.377(3)n4—c131.343(2)c12—c111.391(2)c14—c101.412(2)c2—n11.290(3)n3—c91.353(2)c2—c31.473(2)n3—c61.424(2)c11—c101.385(3)c9—c101.503(2)c4—c31.389(3)c6—c51.392(3)c3—c81.393(3)c6—c71.392(3)c8—c71.386(2)表6式(ⅰ)化合物晶体的部分键角(°)结合附图4、附图5和附图6,表明实施例4所合成的化合物为式(ⅰ)所示化合物。实施例7式(ⅰ)所示化合物的体外抗肿瘤活性选取hepg2肝癌细胞,结肠癌细胞lovo细胞,人非小细胞肺癌a549细胞;人乳腺癌mcf-7,sk-br-3细胞,人宫颈癌hela细胞为测试细胞株。采用mtt法对式(ⅰ)所示化合物进行体外抗肿瘤活性评价。样品用dmso溶解后,然后用dmem培养基稀释成50、20、10、5、1、0.5、0.1、0.01μmol/l不同浓度。取对数生长期的测试细胞株悬浮于含10%胎牛血清的无酚红dmem培养基中,铺至96孔细胞培养板中,每孔加入100μl(4~10)×104个/ml细胞悬液。待细胞完全贴壁后,弃去原培养液,加入100μl的含有测试药物的培养液培养3天后,每孔加入30μl5mg/mlmtt,置于37℃、5%co2培养箱中继续孵育4h,然后每孔加入100μl二甲亚砜(dmso)溶解。使用酶标仪在490nm波长测定每孔的吸光度(od)值,计算被测药物对肿瘤细胞的抑制率(%),采用非线性回归模型绘制s型剂量效应曲线,用originpro软件计算出半数抑制浓度(ic50)值,结果见表7。细胞抑制率(%)=(正常od值-加药od值)/正常od值×100%表7式(i)所示化合物抗肿瘤活性(n=3)测试结果表明,测试样品式(ⅰ)所示化合物对于六种肿瘤细胞均具有明显的抑制活性,尤其是对于hepg2肝癌细胞具有较好抑制活性,ic50=0.5μm,说明该结构具有潜在抗肿瘤活性,可用作抗肿瘤活性先导化合物。实施例8式(ⅰ)所示化合物的体内抗肿瘤活性在体外活性中,式(ⅰ)所示化合物对肝癌hepg2细胞展现出了优秀的抑制活性,因此其进一步进行体内动物实验研究。hepg2细胞(2×106)接种于5周龄(体重15-16g)的balb/c雌性裸鼠左前肢下面,待肿瘤长至60cm3时开始进行体内治疗实验,实验室(n=4)按照25mg/kg的剂量(150μl)灌胃给药,每隔一天给药一次,对照组(n=4)在同等条件下给予等剂量的pbs。给前和给药后每隔2天测量肿瘤的大小和体重,并在治疗结束后取下肿瘤称量其重量。肿瘤的体积(mm3)=0.5×(肿瘤的长度×宽度2)。式(ⅰ)所示化合物的体外抗肿瘤结果如附图7、附图8、附图9和附图10。测试结果表明,式(ⅰ)所示化合物在动物体内能够明显的抑制肿瘤的生长,并且不影响实验室动物的体重,说明该结构不仅具有优秀的体内抗肿瘤活性,而且毒性较低。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1