烷烃磺酸的生产方法与流程
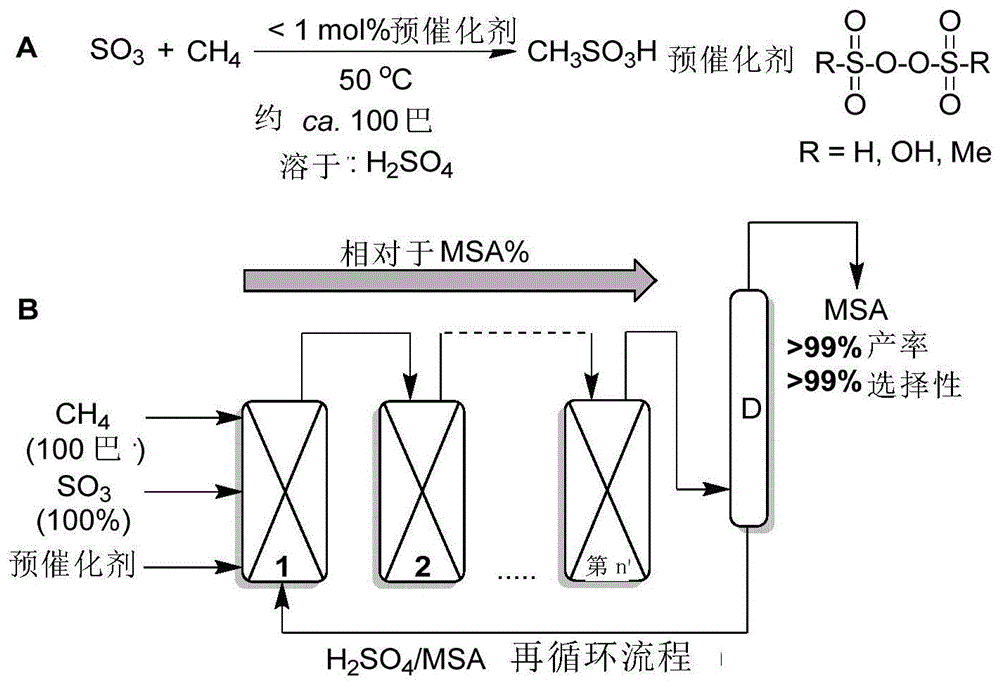
烷烃磺酸是有机酸,可以达到与无机矿物酸(例如硫酸)相似的酸强度。然而,与通常的矿物酸如硫酸和硝酸对比,磺酸是非氧化性的,并且不会如盐酸和硝酸所观察到的那样释放出对健康有害的蒸气。进一步,多种磺酸例如甲磺酸,是可生物降解的。磺酸的应用多种多样,例如用于清洁剂、表面活性剂、催化剂,以及在有机合成、药物化学中的应用,如作为保护基团。磺酸盐,如十二烷基磺酸钠,例如用作表面活性剂,或者在电镀工业中,尤其是作为锡、锌、银、铅和铟以及其它金属的烷基磺酸盐。具体地,烷基磺酸盐的非常高的溶解度起重要作用。此外,在电解中不会形成有害气体,并且省去了有毒化合物(例如在许多情况下常见的氰化物)的使用。结构上最简单的烷烃磺酸代表是甲烷磺酸。水力压裂技术和沼气生产的进步提供了大量廉价甲烷。尽管甲烷储量丰富,但甲烷在转变为更复杂的分子的活化和功能化方面上具有高度惰性,而且最重要的是由于其分子稳定性高,甲烷对气候变化的影响高于二氧化碳。在过去的几十年里,为克服这一挑战做出了非凡的努力,并且已经开发出各种潜在的解决方案。然而,这些方案的工业适用性在某种程度上受到经济约束(例如,昂贵的催化剂、不切实际的可扩展性以及其他问题)的限制。此外,能够升级CH4的处理必须稳健且稳定,以便以工业规模处理大量材料。将甲烷液化为甲醇也是不能实行的,由于合成气生产的CO:H2比例为1:3(甲醇的合成需要CO:2H2混合物)。几种同类的方法使用如Hg,Pt,Ir,Rh,Ti和Pd等过渡金属络合物靶向甲烷的C-H活化。然而,对更复杂分子的功能化仍然是难以掌握的。可以强调三种有趣的功能化方法:1)甲醇;2)硫酸氢甲酯(MBS);和3)甲磺酸(MSA)。甲醇的直接合成具有挑战性,因为甲醇会快速过氧化成更稳定的产品,并且需要严苛的条件。在高温下Hg介导的在H2SO4介质中活化CH4的亲电子C-H的方法产生SO2和MBS,而后者可能在高稀释度下转化为甲醇。关于甲烷磺化的早期工作引发对用于在发烟硫酸中将CH4活化为甲磺酸(MSA)的几种自由基引发剂和添加剂的报道。然而,这些反应缺乏高产率和良好的选择性。此外,反应机制尚未完全了解。扩大生产会出现明显的问题,例如SO2转化的额外催化途径、过大的反应器和极高的温度。与硫酸氢甲酯(MBS)相比,甲磺酸(MSA)被认为是高附加值产品和绿酸(例如,非氧化剂,低蒸气压,可生物降解等),可用于制药、电子和清洁行业。Basickes等人(Basickes,Hogan,Sen.美国化学会期刊(J.Am.Chem.Soc.)1996,11,13111-13112)描述了在发烟硫酸中自由基引发甲烷和乙烷的过程。需要90℃以上的温度,且MSA的收率低。Mukhopadhyay和Bell(有机反应研究与发展(OrganicProcessResearch&Development)2003,7,161-163)报道了在过氧化二磷酸钾作为引发剂存在下,甲烷在低压下直接磺化成甲磺酸。为了活化引发剂,选择95℃的温度并且SO3的转化率低于30%。Lobree和Bell(工业与工程化学研究(Ind.Eng.Chem.Res.)2001,40,736-742)也研究了K2S2O8引发的甲烷磺化成甲磺酸。描述了一种自由基机理,并且需要高温以及低浓度的引发剂以实现适度的SO3转化率。US2,493,038描述了由SO3和甲烷制备甲磺酸。US2005/0070614描述了制备甲磺酸的其他方法及其应用。现有技术中已知的方法相对复杂,成本高,并且由于严苛的条件而导致不合需求的产物。WO2004/041399A2和US7,119,226B2都提出了用于产生甲磺酸的自由基途径和链式反应。大体上,自由基链式反应通常会产生不合需求的副产物,副产物甚至表现为烷烃磺酸生产中的干扰抑制剂,这可能导致制备烷烃磺酸的实际反应终止并进一步导致杂质、形成副产物以及三氧化硫和甲烷的低产率。因此,本发明的目的是提供一种将烷烃增值为烷烃磺酸的工业方法。本发明的一个具体问题是提供这种将甲烷增值为甲磺酸(MSA)的方法。该过程应该避免副产物并且与起始物等摩尔,在绿色化学的十二个原则的意义上是可持续的,并且在经济上是可行的。在第一个实施方案中,该问题通过一种由烷烃(尤其是甲烷)和三氧化硫制备烷烃磺酸(尤其是甲磺酸)的方法来解决,该方法包括使烷烃的碳正离子(尤其是甲烷的碳正离子)与三氧化硫反应形成烷烃磺酸的步骤。令人惊奇地发现,当使用烷烃的碳正离子时,可以获得高收率的烷烃磺酸并且几乎没有任何不合需求的副产物。所述创造性的方法与现有技术的方法的不同之处尤其在于,使用离子途径来制备烷烃磺酸。因此,所述创造性的方法避免了自由基链反应的参与,其通常导致几种不同副产物的形成。在本发明所述方法中未观察到这些副产物。此外,反应可以在需要高温的现有技术中所观察到的不发生自由基链反应的温度下进行。例如,所述创造性的方法可以在室温或约30℃有效地进行。所述创造性的方法中不需要添加促进引发剂分解成自由基或稳定所述自由基的物质。具体地是,在本发明的一个实施方案中没有添加此类物质。此类物质包括金属盐(例如Pt、Hg、Rh)。它们表现出引发副反应的有害副作用,而这种副作用可以通过本发明避免。衍生碳正离子的烷烃可以是任何烷烃,优选甲烷、乙烷、丙烷、丁烷、异丙烷或异丁烷。特别优选地,在本发明方法中使用甲烷作为烷烃以生产甲磺酸。通过这种方式,甲烷可以被增值并且可以很好地利用。本发明所述的碳正离子是具有带+1正电荷的碳离子的任何离子。具体地,碳正离子可以从烷烃衍生出来,通过将H+形式加成到烷烃的碳原子上,形成带正电的五价碳原子,在此表示为阳碳离子(carboniumion)。如果烷烃是甲烷,则相应的阳碳离子是阳甲烷离子(methanium)(CH5+)。碳正离子可以通过形式消除烷烃碳原子的H-而从烷烃得到,继而得到带正电荷的三价碳原子,在此表示为正碳离子(carbeniumion)。如果烷烃是甲烷,则相应的正碳离子是正亚甲基离子(methenium)(CH3+)。本发明方法的下文更详细地描述了优选实施方案和所涉及的离子途径,尤其是关于甲烷转化为甲磺酸。描述涉及甲烷和甲磺酸,并不意味着将本发明的范围限制为甲烷。如作适当变动,相同的考虑适用于其他烷烃的使用,这也在本发明的范围内。选择甲烷仅作为说明性实例,尽管它作为烷烃也是优选的。通过用烷基取代基取代氢原子,甲烷可以转化为其他烷烃。在本发明方法的一个优选实施方案中,碳正离子是正碳离子,尤其是正亚甲基离子(CH3+)。优选地,烷烃的碳正离子与三氧化硫反应的步骤包括以下步骤:i)使正碳离子与三氧化硫反应,形成烷基亚硫酸盐阳离子;ii)使烷基亚硫酸盐阳离子与烷烃反应,形成烷烃磺酸,并再生正碳离子。例如,本发明方法可包括形成CH3+,然后与SO3反应,形成H3C-O-SO2+,随后H3C-O-SO2+与存在于反应混合物中的烷烃反应,从而形成烷烃磺酸,如下图所示:在产生一定量的正碳离子之后,这些离子因此可以用于催化循环,其中三氧化硫与烷烃直接反应得到烷烃磺酸通过离子途径发生。正碳离子可以通过现有技术中已知的任何合适的方法产生。具体地,正碳离子可以通过使烷烃与式R3-O-O-R4的活化预催化剂反应获得,其中该预催化剂包含过氧化氢衍生物,并且其中通过预催化剂与超酸反应而活化预催化剂。超酸是一种酸度大于100%纯硫酸的酸,其哈米特酸度常数H0值为-12。因此,超酸作为一种介质,其中质子的化学势高于纯硫酸中。超酸包括三氟甲磺酸(CF3SO3H)(也称为三氟甲磺酸),和三氟硫酸(HSO3F)或二硫酸(H2S2O7)。如本发明方法所述,优选二硫酸作为超酸。可以通过过量的SO3与硫酸反应来获得二硫酸。R3-O-O-R4可以是任何适合用超酸活化并与烷烃反应形成正碳离子的有机或无机过氧化物。独立于有机或无机,R3和R4可以彼此相同或不同。适合的过氧化物的示例是过氧碳酸酯、过氧磷酸酯、过氧硫酸酯等。优选地,预催化剂对应于下式其中R1和R2可以相同或不同,且选自-H、-OH、-CH3、-O-CH3、-F、-Cl、-Br、-C2H5或更高级的烷烃基、-O-C2H5或更高级的烷烃基的组。特别地,预催化剂可以通过过氧化氢与磺酸(尤其是甲磺酸)反应获得。一种特别合适的预催化剂是不对称单甲基磺酰基过氧化物(MMSP),MMSP可通过甲磺酸与过氧化氢反应得到,任选地在H2SO4中。可通过将前体物质加入反应混合物中原位制备预催化剂。预催化剂通过其与超酸反应来活化。然后,活化的预催化剂与烷烃反应形成正碳离子。优选地,基于三氧化硫的量,预催化剂的用量为0.01mol%至30mol%。更优选地,预催化剂的量为0.5mol%至5mol%。具体地,基于三氧化硫的量,预催化剂可以以0.9mol%的量使用。例如,上述途径具体描述如下,从甲烷形成正亚甲基离子(CH3+)的过程:然而,本发明不限于该途径。优选地,碳正离子的形成,具体地是正碳离子的形成,涉及与超酸(超强酸)的反应。优选地,烷烃直接或间接地与超酸反应。优选地,超强酸存在于反应混合物中。原则上,合适的过氧化物预催化剂也可以通过均裂分解-O-O-过氧键产生两个-O*基团而作为自由基引发剂反应。不受理论束缚下,假定本发明离子过程中的预催化剂以不同方式分解,即不是通过破坏O-O键而是离子裂解R-O键。将质子加入O-O键后,即形成R-O(H+)-O-R中间体,R-OH+键断裂,其中形成R-OOH,并且R+,其之后与烷烃反应生成正碳离子和R的分解产物。因此,本发明中特别优选的是R1和R2不同,即使用非对称的预催化剂。在这种非对称的预催化剂中,O-O键被极化,这有助于使一个氧原子质子化,从而导致过氧化物的异分裂。这种非对称预催化剂的实例是单甲基磺酰基过氧化物,如下式:根据本发明,三氧化硫可以用作纯SO3(100%SO3)。这避免了制备三氧化硫溶液。这里的反应条件没有添加溶剂。此外,未反应的三氧化硫可以蒸发,避免淬火的必要性。另外,三氧化硫可以用于溶液中,或者作为三氧化物含量为50%(w/w)或更低,或65%(w/w)或更高的发烟硫酸。令人惊奇的是,本发明发现,与同样用于本发明方法的现有技术相反,使用三氧化硫含量65%(w/w)或更高,特别是70%w/w或更高的发烟硫酸,对本发明方法不产生负面影响。甚至可以使用纯三氧化硫(100%(w/w)三氧化硫)。发烟硫酸溶液中的三氧化硫含量,优选在15%(w/w)至60%(w/w)和65%(w/w)至99%(w/w)的范围内,优选为25%(w/w)至60%(w/w)和70%(w/w)至95%(w/w),特别优选为35%(w/w)至55%(w/w)和75%(w/w)至90%(w/w)。SO3含量低于15%(w/w)也会形成烷基磺酸,但反应时间太长,以至于出于经济原因,反应将变得无价值。令人惊奇的是,已经发现SO3含量在60%(w/w)至65%(w/w),反应时间也非常慢,因此从经济学上来讲是无价值的。本发明方法可以在反应器中进行。在反应器中提供纯SO3或含有三氧化硫或发烟硫酸的溶液。在同一反应器中,提供烷烃,尤其是甲烷。对于低沸点的烷烃,必须使用高压反应器。对于戊烷和更高级烷烃,常见的实验室反应器足够。在气态烷烃的情况下,例如甲烷,压力设定为1至150巴。所用烷烃为甲烷时,优选的压力在10至150巴的范围内,更优选的在50至120巴的范围内。在反应器中提供超强酸,例如通过向反应器中加入硫酸,其中二硫酸因SO3的存在而形成。如果使用发烟硫酸作为SO3的来源,则不必再添加超强酸。此外,可以添加如上所述的预催化剂。预催化剂也可以以前体的形式加入,具体地是以过氧化氢和烷基磺酸的形式加入,它们将相互反应,在反应混合物中原位形成预催化剂。反应混合物的温度,控制在0至100℃,优选0至50℃的范围内。该温度下烷烃磺酸(尤其是甲磺酸)形成,取决于作为反应物而提供的烷烃。所得产物烷烃磺酸可以纯化,例如通过蒸馏、结晶、萃取或色谱法纯化。与采用自由基链反应的已知的现有技术方法相比,本发明方法的一个特别的优点是可以选择低于自由基形成温度的温度。优选地,温度低于50℃,更优选低于40℃,特别地低于35℃,极其优选地低于30℃。反应也可以在室温下进行。相对于上述温度范围的优选上限,下限具体可以是室温。特别优选的是采用本发明方法,使用甲烷和三氧化硫生产甲磺酸(MSA),这将在下文中更详细地描述。图1显示了本发明在优选实施方案中用于CH4活化和功能化为MSA(参见图1的A部分)。B部分显示了中试工厂中连续反应器(第1,2和n)如何生产高达20吨/年,然后在D列中蒸馏富集的混合物以获得纯MSA。未观察到副产物,并且H2SO4/MSA流再循环回到反应器1。图2显示了在超强酸条件下正过氧化氢离子(hydrogenperoxoniumion)的形成和甲醇分解成MBS。图3(上部)显示了MSA合成的CH4压力(巴)与时间(小时)测量的反应曲线。插图:区域A的特写。(底部)显示了标准反应与添加痕量SO2作为失活剂之间的反应曲线的比较。图4显示了预测的甲烷活化和功能化的阳离子机理:A)预催化剂通过过氧化物1的质子化激活;和B)生产催化循环,其中正亚甲基离子5通过甲烷的脱氢再生。图5显示了在不同温度下甲基亚硫酸氢盐的歧化作用如何提供MSA(50℃)或MBS(120℃)。本发明的一个具体实施方案包括在约100巴的压力下不同浓度(15-60%)发烟硫酸(SO3/H2SO4)溶液中活化甲烷,其溶液中含有约1mol%包含过氧化物衍生物的预催化剂(图1A)。对于中试工厂的生产,反应可以在连续反应器中进行(图1B)。在第一反应器中加入纯SO3和CH4,然后将反应混合物通入下一个,增加MSA的浓度,直至进行蒸馏反应的第n个反应器。馏出物由纯MSA组成,产率超过99%(基于SO3的初始量)和99%选择性。将包含H2SO4和少量MSA的混合物的剩余溶液送回第一反应器,使得在反应开始时所需的发烟硫酸浓度。该配置允许将本方法扩大到大多数工业生产,大约10,000公吨MSA/年。使用400mL间歇式反应器进行反应机理和反应优化的详细研究(表1)。甲烷向混合物中的扩散是转化为MSA的关键,必须使用具有气体扩散功能的螺旋桨。表1显示了使用原位形成的0.9mol%预催化剂MMSP情况下,MSA在不同条件(例如温度,压力等)下的产率。最常见的实验是用34%发烟硫酸进行的,在50℃下16小时内提供60%的MSA产率。使用大型反应器,MSA的产率显著增加到99%。条目3描述了使用UV辐射(见下文)的反应,没有显著的MSA产生。SO2作为失活剂的影响如条目4所示,其中MSA的总收率为23%。表1.在400mL间歇式反应器中合成MSA条目SO3(%)温度(℃)CH4压力(巴)MSA产率(%)a13450976022450978332425960b434509523c53650恒定d97636409626ea基于SO3的初始量,MSA的百分比产率(通过离子色谱法分析);b带紫外线;c加入SO2;d恒定的甲烷压力;e基于反应的甲烷量的转化。具体地,上表的条目3显示该反应通过离子途径发生。即使在25℃的温度下,UV光也会引发自由基链式反应。UV光能够均裂分裂过氧键,导致自由基的形成。不受理论束缚下,假设所述均裂分裂实际上确实发生并使离子途径失效,这可能涉及如上所述的R1-O-O-R2的异分裂。对不同组成的预催化剂进行研究。例如,H2SO4(98%)中只含有H2O2(60%)不会显著触发MSA的形成。在超强酸条件下,H2O2形成正过氧化氢离子H3O2+,其与CH4反应,继而提供甲醇(图2)。H2O2,MSA和H2SO4的混合物表现出最佳的MSA的合成性能。在预催化剂混合物中鉴定出不对称的单甲基磺酰基过氧化物(MMSP)。现有技术中已知对称的二甲基磺酰基过氧化物(DMSP)能够产生显著的MSA产率,然而,该预催化剂选择性和速率更优。通过NMR,IR和MS鉴定MMSP。获得以下信号:1HNMR(纯H2SO4,毛细管CDCl3):δ12.23(ovs),5.38(ovs)。FT-IR(ATRcm-1):1693(S-O),1334(S=O)。ESI-MS(m/z):192.86(MH-)。标准反应的反应曲线(发烟硫酸30%,原位制备的0.9mol%MMPS,大约100巴甲烷,反应器加热至50℃)显示在加入预催化剂后长达2小时的特定时间内,压降几乎是线性的(图3A)。之后,压力的降低类似于快速衰减(图3B),然后在10小时后达到平台(图3C)。基于压降的MSA的计算产率和离子色谱分析表明,在诱导期内形成了相对于H2O2的摩尔数的等摩尔量的产物(图3A)。此外,通过氧化还原滴定测量预催化剂在50℃和大气压下的分解,在最初的70分钟内迅速发生40%过氧化物的分解。这表明上述时期可能确实是催化活化(图3A)。该反应对温度高度敏感,影响多种产物分布并显著影响比率。低温(低于50℃)提供高于99%MSA的选择性,然而,在较高温度(高于100℃)下,反应开始表现出更复杂的产物混合物,其中MBS和SO2为主要组分。在反应7天时间后,在20℃下可以获得高达85%的MSA产率。另一方面,类似于随温度变化观察到的,SO3的浓度对MSA的选择性具有重要影响。在浓度高达60%的SO3时,MSA的产量是定量的,相反,更高浓度的SO3(>60%)促进了MBS和SO2的形成,同时降低了MSA的产率和选择性。乙烷、SO2和O2作为失活剂也具有重要作用。例如,浓度分别为1.29%和0.44%(基于SO3的总量)的SO2和C2H6,能完全淬灭MSA的合成。不同发烟硫酸的哈米特酸度值H0随SO3的量而增加。例如,35mol%的发烟硫酸的H0值为-13.94,其酸度的增加与SO3的浓度一致,然而,在较高值超过50mol%SO3时,酸度的增加非常小(例如,对于75mol%H0=-14.96)。这种趋势与MSA合成中的观察结果一致,其中在较高SO3含量下,MSA的选择性随着MBS和SO2大量形成而降低。Olah和其同事(Olah,Prakash,Sommer,Molnar,超强酸化学(SuperacidChemistry),威利国际科学出版社(Wiley-Interscience),2版2009)已广泛证明,在超强酸条件下(例如,H2SO4的发烟硫酸),溶解的甲烷被质子化到2e-3cCH5+五配位阳离子。类似地,H2O2被质子化以产生高活性正过氧化氢离子H3O2+,其已经被用于大量转化。在大气压下,在D2SO4中CH4的快速H/D交换也证明了在临界超强酸中CH4的C-H容易活化。测试了在超强酸条件下自由基可能参与MSA的形成。在有和没有添加预催化剂的情况下,在室温下用宽波长汞灯照射紫外光不足以触发98巴的CH4形成MSA。另一方面,对照实验表明,原位制备的0.9mol%MMSP预催化剂能够在25℃下在UV照射下聚苯乙烯。相反,使用相同的预催化剂混合物在没有UV光的情况下未观察到苯乙烯的聚合。基于所描述的观察,甲烷的正离子活化,然后在超强酸条件下功能化MSA的过程,如图4所示。如图4所示,MMSP1最初质子化为过氧化阳离子2,随后产生以氧和硫为中心的阳离子(4a或4b),而羟基过氧化物3在与过量SO3反应时形成另一分子MMSP1(方案1A)。物质4a或4b通过提取亲电子氢化物活化CH4以形成正亚甲基离子5。需着重关注的是MMSP1的催化剂量,约是基于SO3的总量的0.9mol%,对应于CH3+进入图4B中的生产催化循环的剂量。SO3亲核攻击CH3+,产生以硫和氧基为中心的甲基亚硫酸盐阳离子(6a和6b),其与在预催化剂活化循环中产成的那些类似。甲基亚硫酸盐阳离子6b可与CH4反应生成甲基亚硫酸氢盐7,其在50℃下快速重排成MSA。一旦预催化剂MMSP1被完全消耗,生产催化循环通过形成CH3+5进行自催化。假定的催化循环考虑了图3中观察到的三个不同时期的反应曲线(参见上文)。图5显示了不同温度下甲基亚硫酸氢盐的重新排列。高温引发异构化为SO2和甲醇,后者随即与游离SO3反应生成MBS。从反应混合物中分离MSA具有挑战性。真空分离实现了高纯度的MSA,然而,高温产生了例如无水甲磺酸和甲磺酸酯的分解产物。在此条件下,MSA的分解产物未见MBS。将蒸馏步骤合并到连续式反应器中;本发明提供了仅用两种反应物SO3和CH4大量生产MSA的高效方法。在另一个实施方案中,为实现本发明的目的,通过使用碳正离子制备链烷(或烷烃)磺酸,尤其是甲磺酸。优选地,碳正离子是正碳离子,尤其是正亚甲基离子(CH3+)。特别地,碳正离子可用于以甲烷和三氧化硫生产甲磺酸。在另一个实施方案中,为实现本发明的目的,通过一种链烷磺酸(尤其是甲磺酸)的制备方法,包括以下步骤:i)提供三氧化硫;ii)提供烷烃,尤其是甲烷;iii)提供预催化剂,其中预催化剂包含过氧化氢衍生物;iv)通过与超强酸(尤其是在H2SO4中的SO3)反应来活化预催化剂;v)在50℃或更低的温度下,使预催化剂、三氧化硫和烷烃在反应器中反应;vi)从反应混合物中分离烷烃磺酸(尤其是甲磺酸)。优选地,在本发明方法中不使用促进自由基形成或使其稳定的物质。具体地,未加入金属盐到反应混合物中。优选地,预催化剂对应于式R1-O-O-R2,其中R1和R2不同,并且视需要预催化剂中的过氧键为极化的。更优选地,预催化剂对应于下式其中R1和R2可以相同或不同,并且选自-H、-OH、-CH3、-O-CH3、-F、-Cl、-Br、-C2H5或更高级的烷烃、-O-C2H5或更高级的烷烃的组。预催化剂可以在步骤iii)中通过提供过氧化氢、烷烃磺酸(尤其是甲烷)和硫酸的混合物提供。优选地,步骤v)中的温度为40℃或更低,更优选地是30℃或更低,特别优选地是25℃或室温。在本发明方法的一个优选实施方案中,步骤v)中的压力在10至150巴的范围内,优选地是在50至120巴的范围内。三氧化硫可以纯三氧化硫形式或在发烟硫酸中的三氧化硫溶液形式使用,具体地是以发烟硫酸中15-60%的三氧化硫溶液形式。实施例实施例1:MSA的合成步骤在400mL不锈钢高压反应器中,使用HPLC泵加入245.02g发烟硫酸(34.1%),同时保持管线温度为50℃。将反应器加热至50℃,搅拌速度恒为1000rpm。在12mL硫酸(98%)和1.38mLMSA(99.5%)的冷混合物(0℃)中逐滴加入464μL过氧化氢(60%),以制备预催化剂。一旦反应器达到50℃的恒定温度,用92.6巴甲烷(99.5%)加压容器。然后使用HPLC泵将预催化剂注入反应器中,将反应器内的压力升至97巴。16小时后,压力降至31.8巴,表明大量甲烷被消耗。然后将反应器冷却至室温,将过压的甲烷再次移至洗涤器中,将无色液体的样品储存在玻璃瓶中,称重为279.57g。随后使用IC分析样品,基于SO3的总初始摩尔数,得到MSA产率为59.9%。实施例2:MSA的合成步骤在400mL不锈钢高压反应器中,使用HPLC泵以恒定温度50℃加入288.07g发烟硫酸(24%)。然后将反应器加热至50℃并将搅拌器设定为1000rpm。一旦反应器内的温度恒定在50℃,就加入92.6巴的甲烷。在12mL硫酸(98%)和1.38mLMSA(99.5%)的冷混合物(0℃)中逐滴加入464μL过氧化氢(60%)来单独制备预催化剂。使用HPLC泵将预催化剂加入反应器中,使总压力增至97.4巴。反应20小时后,压力降至26.1巴。然后将反应器冷却至室温,并将过量的甲烷转移至洗涤器中。将反应器内的全部内容物(264.2g)转移到玻璃瓶中并适当储存。IC分析表明,基于加入的SO3总量,该反应中MSA的产率为83.3%。实施例3:紫外线照射尝试合成MSA的步骤(对比例)在配有两个长方形蓝宝石窗口的400mL不锈钢高压反应器中,使用维持在50℃的HPLC泵添加249.31g发烟硫酸(24%)。然后将反应器加热至25℃,将搅拌器设定为1000rpm并用91.7巴的甲烷加压。一个中等压力190nm的宽波发射,配有石英浸入管和冷却夹套的汞蒸汽紫外灯(UV-ConsultingPeschl,德国),置于离反应器窗口4厘米处,都覆盖铝箔。紫外灯产生的热量不足以改变反应器内的温度。在12mL硫酸(98%)和1.38mLMSA(99.5%)的冷混合物(0℃)中逐滴加入464μL过氧化氢(60%)以制备预催化剂。然后使用HPLC泵将预催化剂加入反应器中,达到95.7巴的总压力。2小时后,反应器内的温度保持稳定在25℃,压力恒定在96巴。在4小时的反应时间内,压力仍然保持在95.9巴。压力值不变表明甲烷没有消耗,并且在这些条件下不产生MSA。实施例4:MSA的合成步骤在4L不锈钢高压反应器中,通过套管加入1.943kg发烟硫酸(36%)。将反应器保持在40℃,搅拌速度为350rpm。一旦温度恒定,向反应器中加入95.6巴甲烷。单独地,通过在90mL硫酸(98%)和10mLMSA的冷混合物(0℃)中逐滴加入3.4mLH2O2(70%)来制备预催化剂。使用HPLC泵将预催化剂加入反应器中,将总压力提高至98.5巴。在实验期间不断监测压力。反应16小时后,压力为71.1巴。反应67小时后,反应器内的压力降至31.1巴。此时冷却反应器,将过量压力的甲烷移到一组充满硫酸的洗涤器中并取出样品。将样品储存在玻璃瓶中,称重为2.244kg。计算出甲烷在16小时的转化率(基于SO3的初始量)为26%。离子色谱分析表明,67小时后MSA的产率为92%MSA。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1