用于显示器基底的聚酰亚胺膜的制作方法
本申请要求于2017年11月21日提交的韩国专利申请第10-2017-0155408号的优先权的权益,其全部公开内容通过引用并入本文。
本发明涉及用于柔性显示器基底的聚酰亚胺膜,其可以使在tft工艺期间形成在聚酰亚胺基底上的无机膜层中的裂纹的产生最小化。
背景技术:
近来的显示装置集中于重量减轻和使其小型化。例如,用于显示装置的玻璃基底具有重且容易破裂而难以连续处理的限制,并且已经被具有轻质、柔性并且能够在诸如移动电话、笔记本电脑、pda等的装置中连续处理的优点的塑料基底替代。
特别地,聚酰亚胺(pi)树脂具有其易于合成,并且其可以以薄膜的形式制备并且不需要用于固化的交联基团的优点。由于这些原因,根据近来电子产品的趋势例如轻质和精细化,作为lcd、pdp等的半导体用集成材料,许多研究人员试图将pi用于具有轻质和柔性特性的柔性塑料显示板。
通常,聚酰亚胺树脂以膜的形式制备,特别地通过二酐和二胺或二异氰酸酯的溶液聚合以获得聚酰胺酸衍生物的溶液并将该溶液涂覆在硅晶片或玻璃上,然后进行热固化来制备。
技术实现要素:
技术问题
设计本发明以解决相关技术的技术问题,因此,本发明的一个方面是提供聚酰亚胺膜,其可以使待形成在膜上的无机膜层中的裂纹的产生最小化。
本发明的其他方面是提供由聚酰亚胺膜制备的柔性显示器基底。
技术方案
为了实现以上方面,
本发明提供了用于柔性显示器基底的聚酰亚胺膜,其具有5μm至20μm的厚度,并且当其在硅晶片上在350℃至500℃的温度下经受加热和冷却时具有0至5的应力变化。
在一个实施方案中,聚酰亚胺膜可以通过使用包含聚酰亚胺前体和具有正的logp值的有机溶剂的溶液来制备。
在一个实施方案中,具有正的logp值的有机溶剂可以为选自以下的至少一者:二甲基丙酰胺(dmpa)、二乙基丙酰胺(depa)、n,n-二乙基乙酰胺(deac)、n,n-二乙基甲酰胺(def)和n-乙基吡咯烷酮(nep)。
在一个实施方案中,具有正的logp值的有机溶剂的密度可以为1g/cm3或更小。
在一个实施方案中,具有正的logp值的有机溶剂的蒸气压可以为0.5托或更大。
在一个实施方案中,聚酰亚胺膜在350℃至500℃的温度下可以具有正的cte值。
在一个实施方案中,聚酰亚胺可以包括使用二胺、酸二酐和封端剂作为聚合组分制备的封端的聚酰亚胺。
在一个实施方案中,聚酰亚胺可以具有下式6的重复单元:
[式6]
在式6中,
r1、r2和r3各自独立地选自氢原子、c1-10烷基、c1-10氟烷基、c6-12芳基、羟基和羧基。
在一个实施方案中,聚酰亚胺还可以具有下式6a的重复单元:
[式6a]
在式6a中,
r4、r5、r6和r7各自独立地选自氢原子、c1-10烷基、c1-10氟烷基、c6-12芳基、羟基和羧基。
在一个实施方案中,在式6a中,r4和r5可以各自独立地为氢原子,以及r6和r7各自独立地为c1-10氟烷基。
本发明还提供了由聚酰亚胺膜制备的柔性显示器基底。
有益效果
本发明提供了聚酰亚胺膜,当将其涂覆在玻璃基底上时,其在高温下的应力变化低。聚酰亚胺膜可以使由于在通过在聚酰亚胺基底上沉积形成无机膜的过程期间由聚酰亚胺膜的应力变化引起的无机膜的变形而引起的裂纹产生最小化,并因此可以减少柔性显示器的可恢复的残像和电流减小。
附图说明
图1示出了对于实施例1中制备的膜测量的残余应力的结果。
图2示出了对于实施例1和比较例1中制备的膜测量的cte变化和残余应力变化的结果。
图3示意性地示出了根据在实施例1和比较例1中制备的膜上沉积无机膜层的应力行为。
具体实施方式
在下文中,将参照附图详细地描述本发明,附图示出了本发明的优选实例用于更好地说明的目的,而不旨在限制本发明的技术范围。对于本领域技术人员明显的是,在不脱离本发明的精神和范围的情况下,可以做出各种改变和修改。在以下描述中,如果公知的功能或结构可能使本发明的观点模糊,则将不对其进行详细描述。
如本文所用,除非另有说明,否则所有化合物或有机基团可以为经取代或未经取代的。术语“取代”意指化合物或有机基团中包含的至少一个氢经以下取代:卤素原子、具有1至10个碳原子的烷基、卤代烷基、具有3至30个碳原子的环烷基、具有6至30个碳原子的芳基、具有1至10个碳原子的烷氧基、羧基、醛基、环氧基、氰基、硝基、氨基、及其衍生物。
目前,已经使用塑料基底制备显示器以减小玻璃基底的重量和厚度。特别地,在塑料基底上施加有oled器件的显示器具有弯曲或折叠的优点,并且正在持续地被开发。
这样的柔性显示器装置通过在tft器件的制备中在固化的聚酰亚胺上形成缓冲层、有源层(activelayer)和栅极绝缘体的多层无机膜来制备。作为无机膜,主要使用硅氧化物膜(siox)或硅氮化物膜(sinx)。公知siox在工艺中具有被压缩的特性,sinx具有拉伸特性。因此,当聚酰亚胺基底在用于在基底的tft工艺期间沉积无机膜的高温下经受加热时,需要使聚酰亚胺基底的应力变化最小化,从而使由于在形成无机膜时聚酰亚胺膜的热变形而引起的无机膜中的裂纹的产生最小化。聚酰亚胺基底的这些特性与关于制备柔性oled时柔性显示器的可恢复的残像和电流减小的电特性密切相关。
为了解决相关技术的问题,
本发明提供了用于柔性显示器基底的聚酰亚胺膜,其具有5μm至20μm的厚度,并且当其在硅晶片上在350℃至500℃的温度下经受加热和冷却时具有0至5的应力变化。
本发明使用在350℃至500℃的范围内具有0至5的应力变化的聚酰亚胺膜以使在用于沉积无机膜的高温过程期间由应力引起的聚酰亚胺的变化最小化,从而抑制在聚酰亚胺膜上沉积无机膜时产生的弯曲现象并且减少应力反向(stressreversal)的发生。
在一个实施方案中,聚酰亚胺膜可以具有5μm至10μm的厚度,并且在350℃至500℃的温度下具有0至3的应力变化。
如果聚酰亚胺膜由于热收缩而具有过量的应力变化,则与无机膜层的应力反向的发生可能增加,从而在无机膜中产生裂纹。由于这个原因,优选地,聚酰亚胺膜的应力变化为0至3。
此外,根据本发明的聚酰亚胺膜在350℃至500℃的温度下可以具有正的cte(热膨胀系数)值。例如,cte值可以为0ppm/℃至10ppm/℃,优选为0ppm/℃至5ppm/℃。这表示根据本发明的聚酰亚胺膜在如上所述的高温下不经历收缩,从而抑制由热收缩引起的聚酰亚胺的弯曲,然后抑制应力反向的发生。
因此,根据本发明的聚酰亚胺膜在高温过程中具有低的应力变化和正的cte值,从而减少由于在沉积无机膜的过程中的残余应力而引起的与无机膜层的应力反向的发生,然后使无机膜层中的由于这样的残余应力引起的裂纹的产生最小化。即,在沉积用于制备tft器件的siox或sinx层时,可以使由于聚酰亚胺与siox层的应力反向引起的裂纹的产生最小化,并且减少柔性显示器的tft器件中可能由裂纹引起的可恢复的残像和电流减小。
根据本发明的用于聚酰亚胺前体的聚合的有机溶剂可以具有正的分配系数(logp)值。
有机溶剂可以具有在25℃下的正分配系数(logp)和180℃或更小的沸点。更优选地,分配系数(logp)值可以为0.01至3、或0.01至2。
分配系数可以使用acd/percepta平台(acd/labs)的acd/logp模块来计算。acd/logp模块使用用于基于分子2d结构的定量结构-特性关系(qspr)的算法。
具有正分配系数(logp)的溶剂可以为基于酰胺的溶剂,其可以为选自以下的至少一者:二甲基丙酰胺(dmpa)、二乙基丙酰胺(depa)、n,n-二乙基乙酰胺(deac)、n,n-二乙基甲酰胺(def)和n-乙基吡咯烷酮(nep)。其中,二甲基丙酰胺(dmpa)或二乙基丙酰胺(depa)在以下方面是最优选的:相对低的极性和低沸点以提供良好的涂覆特性以及在低温下的优异的挥发性,这可以降低在膜形成之后残留在膜中的溶剂的量。
正分配系数值意指溶剂的疏水极性。本发明人发现,当使用具有正分配系数的特定溶剂来制备聚酰亚胺前体组合物时,可以改善溶剂的干燥特性(dryingproperty)。此外,使用具有正分配系数的溶剂可以在不使用用于控制涂覆膜的表面张力和光滑度的添加剂例如流平剂的情况下控制溶剂的干燥特性。通过避免使用另外的添加剂,不仅可以消除最终产品中包含低分子量材料而引起品质损坏和加工困难的问题,而且还具有更有效地形成具有均匀特性的聚酰亚胺的效果。
例如,在将聚酰亚胺前体组合物涂覆在玻璃基底上的过程中,当涂覆溶液固化或在湿度条件下储存时,涂覆溶液可能由于涂层的收缩而反润湿。涂覆溶液的这种液体卷曲现象可能引起膜厚度的变化,从而由于缺乏膜的耐弯曲性而导致膜断裂或在切割时边缘开裂的现象,因此产生可加工性差和产率降低的问题。
此外,当可能向涂覆在基底上的聚酰亚胺前体组合物中引入细小的极性异物时,包含具有负logp值的极性溶剂的聚酰亚胺前体组合物可能散发性地经历由涂覆膜中的异物的极性引起的基于异物的位置的裂纹产生或厚度变化,而包含具有正logp值的疏水性溶剂的聚酰亚胺前体组合物即使引入了细小的极性异物,也可以减少或抑制裂纹产生和厚度变化。
具体地,包含具有正logp值的溶剂的聚酰亚胺前体组合物如由以下方程式2所定义的反润湿率可以为0%至0.1%:
[方程式2]
反润湿率(%)=[(a-b)/a]×100
在方程式2中,
a为在聚酰亚胺前体组合物完全涂覆在基底(100mm×100mm)上的状态下的聚酰亚胺前体组合物的面积,
b为在聚酰亚胺前体组合物或聚酰亚胺膜从涂覆基底的端部反润湿之后的聚酰亚胺前体组合物或聚酰亚胺(pi)膜的面积。
聚酰亚胺前体组合物或膜中的反润湿现象可能在涂覆聚酰亚胺前体组合物之后的30分钟内发生。特别地,随着从边缘开始反润湿,边缘的厚度可能增加。
在将根据本发明的聚酰亚胺前体组合物涂覆在基底上,然后在湿度条件下储存10分钟或更长时间,例如10分钟或更长时间,例如40分钟或更长时间之后,涂覆的聚酰亚胺前体组合物的反润湿率可以为0.1%或更小。例如,即使在20℃至30℃的温度下以及在40%或更大的湿度条件(更具体地,40%至80%,即分别为40%、50%、60%、70%和80%的湿度条件,例如50%的湿度条件)下储存10分钟至50分钟之后,反润湿率也可以非常低,为0.1%或更小,优选0.05%,更优选接近0%。
在固化之后也保持这样的反润湿率。例如,在将聚酰亚胺前体组合物涂覆在基底上,将其例如在20℃至30℃的温度下以及在40%或更大的湿度条件(更具体地,40%至80%,即分别为40%、50%、60%、70%和80%的湿度条件)下储存10分钟或更长时间,例如在50%的湿度条件下储存10分钟至50分钟,然后固化,由此获得的聚酰亚胺膜的反润湿率可以为0.1%或更小。换言之,在通过热处理进行的固化过程中反润湿可能很少或者没有反润湿,并且具体地,反润湿率可以为0.05%,更优选接近0%。
根据本发明的聚酰亚胺前体组合物可以解决反润湿现象,从而提供具有更均匀的特性的聚酰亚胺并因此改善制造过程的产率。
此外,可以通过astmd1475的标准测量方法测量根据本发明的有机溶剂的密度,并且密度可以为1g/cm3或更小。如果密度大于1g/cm3,则相对粘度可能增加,然后过程效率可能降低。
此外,有机溶剂的蒸气压可以为0.5托或更大。当蒸气压为0.5托或更大时,可以提高低温下的挥发性,从而降低在膜形成之后残留在膜中的溶剂的量,这对于形成膜是优选的。
在一个实施方案中,聚酰亚胺可以具有下式1的重复单元:
[式1]
在式1中,
x选自衍生自四羧酸二酐的芳族、脂环族和脂族四价有机基团,以及
y选自衍生自二胺的芳族、脂环族和脂族二价有机基团。
在一个实施方案中,聚酰亚胺可以具有下式1a的重复单元:
[式1a]
在式1a中,
x2选自衍生自四羧酸二酐的芳族、脂环族和脂族四价有机基团,以及
y2选自衍生自二胺的芳族、脂环族和脂族二价有机基团。
x和x2各自独立地为选自下式2a至2d的四价有机基团:
[式2a]
[式2b]
[式2c]
[式2d]
[式2e]
[式2f]
[式2g]
在式2a至2g中,
r31至r42各自独立地为c1-10烷基(例如甲基、乙基、丙基、异丙基、叔丁基、戊基或己基);或c1-10氟烷基(例如氟甲基、全氟乙基或三氟甲基),
a1为0至2的整数,b1为0至4的整数,c1为0至8的整数,d1和e1各自独立地为0至3的整数,f1和g1各自独立地为0至4的整数,h1和j1各自独立地为0至3的整数,i1为0至4的整数,k1和l1各自独立地为0至4的整数,
a1、a2和a3各自独立地选自单键、-o-、-cr46r47-、-c(=o)-、-c(=o)o-、-c(=o)nh-、-s-、-so2-、亚苯基、及其混合物,其中r46和r47各自独立地选自氢;c1-10烷基(例如甲基、乙基、丙基、异丙基、叔丁基、戊基或己基);或c1-10氟烷基(例如氟甲基、全氟乙基或三氟甲基),
此外,x和x2各自独立地为选自下式3a至3k的四价有机基团:
式3a至3k的每个四价芳族有机基团中存在的至少一个氢原子还可以经选自以下的取代基取代:c1-10烷基(例如甲基、乙基、丙基、异丙基、叔丁基、戊基或己基);c1-10氟烷基(例如氟甲基、全氟乙基或三氟甲基);羟基和羧基。
y和y2各自独立地为选自下式4a至4d的二价有机基团:
[式4a]
[式4b]
在式4b中,l1为单键、-o-、-co-、-s-、-so2-、-c(ch3)2-、-c(cf3)2-、-conh-、-coo-、-(ch2)n1-、-o(ch2)n2o-、-och2-c(ch3)2-ch2o-或-coo(ch2)n3oco-,其中n1、n2和n3各自为1至10的整数。
[式4c]
在式4c中,l2和l3彼此相同或不同,并且各自为单键、-o-、-co-、-s-、-so2-、-c(ch3)2-、-c(cf3)2-、-conh-、-coo-、-(ch2)n1-、-o(ch2)n2o-、-och2-c(ch3)2-ch2o-或-coo(ch2)n3oco-,其中n1、n2和n3各自为1至10的整数。
[式4d]
在式4d中,l4、l5和l6彼此相同或不同,并且各自为单键、-o-、-co-、-s-、-so2-、-c(ch3)2-、-c(cf3)2-、-conh-、-coo-、-(ch2)n1-、-o(ch2)n2o-、-och2-c(ch3)2-ch2o-或-coo(ch2)n3oco-,其中n1、n2和n3各自为1至10的整数。
此外,y和y2各自独立地为选自下式5a至5k的二价有机基团:
式5a至5k的每个二价芳族有机基团中存在的至少一个氢原子还可以经选自以下的取代基取代:c1-10烷基(例如甲基、乙基、丙基、异丙基、叔丁基、戊基或己基);c1-10氟烷基(例如氟甲基、全氟乙基或三氟甲基);c6-12芳基(例如苯基或萘基);羟基和羧基。
式1和1a的聚酰亚胺可以通过四羧酸二酐和二胺的聚合来制备。酸二酐和二胺可以以0.95:1至1:0.95,优选0.98:1至1:0.98,或0.99:1至1:0.99的摩尔比使用。
在一个实施方案中,聚酰亚胺可以具有下式6的重复单元:
[式6]
在一个实施方案中,聚酰亚胺还可以具有下式6a的重复单元:
[式6a]
在式6和6a中,
r1、r2、r3、r4、r5、r6和r7各自独立地选自氢原子;c1-10烷基(例如甲基、乙基、丙基、异丙基、叔丁基、戊基或己基);c1-10氟烷基(例如氟甲基、全氟乙基或三氟甲基);c6-12芳基(例如苯基或萘基);羟基和羧基。
此外,当在合成本发明的聚酰胺酸或聚酰亚胺时,需要使过量的聚氨基团或酸酐基团失活。为此,还可以添加二羧酸酐或单胺作为封端剂以封端聚酰亚胺的端部。聚酰亚胺优选由二羧酸酐封端以改善耐热性。
基于100mol的总四羧酸二酐或总二胺,封端剂以1mol%至5mol%,优选1mol%至3mol%的量使用。
适用于封端聚酰亚胺或聚酰胺酸的二羧酸酐的实例包括邻苯二甲酸酐、2,3-二苯甲酮二羧酸酐、3,4-二苯甲酮二羧酸酐、2,3-二羧基苯基苯基醚酐、2,3-联苯二羧酸酐、3,4-联苯二羧酸酐、1,2-萘二羧酸酐、2,3-萘二羧酸酐、1,8-萘二羧酸酐、1,2-蒽二羧酸酐、2,3-蒽二羧酸酐和1,9-蒽二羧酸酐。这些二羧酸酐可以在其分子中具有与胺或二羧酸酐不反应的基团。
适用于封端聚酰亚胺或聚酰胺酸的单胺的实例包括苯胺、邻甲苯胺、间甲苯胺、对甲苯胺、2,3-二甲苯胺、2,4-二甲苯胺、2,5-二甲苯胺、2,6-二甲苯胺、3,4-二甲苯胺、3,5-二甲苯胺、邻氯苯胺、间氯苯胺、对氯苯胺、邻溴苯胺、间溴苯胺、对溴苯胺、邻硝基苯胺、间硝基苯胺、对硝基苯胺、邻氨基苯酚、间氨基苯酚、对氨基苯酚、邻甲氧苯胺(o-anilidine)、间甲氧苯胺、对甲氧苯胺、邻乙氧基苯胺(o-phenetidine)、间乙氧基苯胺、对乙氧基苯胺、邻氨基苯甲醛、间氨基苯甲醛、对氨基苯甲醛、邻氨基苄腈、间氨基苄腈、对氨基苄腈、2-氨基联苯、3-氨基联苯、4-氨基联苯、2-氨基苯基苯基醚、3-氨基苯基苯基醚、4-氨基苯基苯基醚、2-氨基二苯甲酮、3-氨基二苯甲酮、4-氨基二苯甲酮、α-萘胺、β-萘胺、1-氨基-2-萘酚、2-氨基-1-萘酚、4-氨基-1-萘酚、5-氨基-1-萘酚、5-氨基-1-萘酚、5-氨基-2-萘酚、7-氨基-2-萘酚、8-氨基-2-萘酚、1-氨基蒽、2-氨基蒽和9-氨基蒽。这些单胺可以在其分子中具有与胺或二羧酸酐不反应的基团。
此外,可以通过以下进行所得聚酰亚胺的进一步封端:通过在四羧酸二酐和二胺的反应之后添加封端剂以进行连续反应,通过在向二胺中添加基于二羧酸酐的封端剂的反应之后添加四羧酸二酐以进行连续反应,通过在向四羧酸二酐中添加基于二胺的封端剂的反应之后添加二胺以进行连续反应,或者通过同时添加四羧酸二酐、二胺和封端剂。
基于100重量份的四羧酸二酐和二胺的总和,封端剂以20重量份或更小,优选1重量份至10重量份,更优选1重量份至5重量份的量使用。
酸二酐和二胺的聚合可以通过聚酰亚胺或其前体的常规聚合方法例如溶液聚合来进行。
聚合反应可以在无水条件下,在-75℃至50℃,优选0℃至40℃的温度下进行。将二胺化合物溶解在有机溶剂中,向其中添加酸二酐。二胺化合物和酸二酐以约10重量%至30重量%的量包含在聚合溶剂中,并且其分子量可以根据聚合时间和反应温度来控制。
可以将通过以上方法获得的聚酰亚胺前体组合物涂覆在基底的一个表面上,然后酰亚胺化,固化并与基底分离,以制备聚酰亚胺膜。
具体地,通过以上方法获得的聚酰亚胺前体组合物可以为聚酰亚胺前体溶解在有机溶剂中的溶液的形式。例如,在聚酰亚胺前体在有机溶剂中合成的情况下,聚酰亚胺前体组合物可以为在聚合之后获得的聚酰亚胺前体溶液本身,可以进一步添加相同的溶液,或者可以在聚合之后用另一溶剂稀释。
考虑到在形成膜时的可加工性例如涂覆特性,聚酰亚胺前体组合物优选具有提供合适的粘度的固体含量。基于聚酰亚胺前体组合物的总重量,固体含量可以为5重量%至20重量%。优选地,聚酰亚胺前体组合物的粘度为400cp至50,000cp。此外,聚酰亚胺前体组合物的粘度可以小于400cp。如果聚酰亚胺前体组合物的粘度超过50,000cp,则其柔性降低,使得难以均匀地涂覆在基底上并在制备显示器基底时引起过程问题。
在将聚酰亚胺前体组合物涂覆在基底的一个表面上之后,使其经受热处理并与基底分离以制备聚酰亚胺膜。
基底可以为玻璃、金属基底或塑料基底,但不特别限于此。其中,就以下方面而言可以优选使用玻璃:其在聚酰亚胺前体的酰亚胺化和固化期间具有良好的热稳定性和化学稳定性,并且其可以容易地与固化之后获得的聚酰亚胺膜分离而没有任何损坏。
涂覆步骤可以通过常规方法进行,具体地,旋涂、棒涂、辊涂、气刀涂覆、凹版涂覆、反向辊涂覆、接触辊涂覆、刮刀涂覆、喷涂、浸涂或刷涂。特别地,就允许连续过程并且可以增加聚酰亚胺膜的酰亚胺化率而言,可以优选浇铸涂覆。
此外,聚酰亚胺组合物可以以使得最终的聚酰亚胺膜可以具有适合于显示器基底的厚度的厚度涂覆。
具体地,其可以以最终的聚酰亚胺膜可以具有10μm至30μm,优选10μm至20μm的厚度的量涂覆。
在涂覆聚酰亚胺前体组合物之后,在热处理之前可以选择性地进行干燥过程以除去残留在聚酰亚胺前体组合物中的溶剂。
干燥过程可以通过常规方法进行,具体地在140℃或更低,或者80℃至140℃的温度下进行。低于80℃的干燥温度可能增加过程时间,高于140℃的干燥温度可能引起急剧的酰亚胺化,使得难以获得均匀厚度的聚酰亚胺膜。
随后,可以在450℃或更高的温度下进行热处理。
此外,热处理可以在200℃至500℃下以多个阶段进行。例如,热处理可以在450℃或更高的温度下进行一次,或者可以以多个阶段进行至少两次。当热处理以两个或更多个阶段进行时,最终的热处理温度可以为450℃或更高。
然后,可以通过常规方法将形成在基底上的聚酰亚胺膜与基底分离以制备聚酰亚胺膜。
本发明还提供了由聚酰亚胺膜制备的柔性显示器基底。
使用聚酰亚胺膜作为显示器基底可以抑制可靠性劣化,例如可能在用于在显示器基底上设置器件的高温过程中产生的涂层的弯曲和提升,并且可以抑制在高温下的tft工艺期间siox层中裂纹的产生。因此,其可以改善关于可恢复的残像和电流减小的电特性,从而提供更可靠的装置。因此,聚酰亚胺可以有效地用于制备应用于电子器件例如oled、lcd、电子纸或太阳能电池中的柔性显示器,特别是用作用于显示器例如oled的基底。
在下文中,将参照实施例更详细地描述本发明。对于本领域技术人员显而易见的是,以下实施例旨在举例说明本发明,并且不应被解释为限制本发明的范围。
<溶剂>
dmpa、depa、dmac、deac和nmp的特性示于表1中。
[表1]
dmpa:n,n-二甲基丙酰胺
depa:n,n-二乙基丙酰胺
dmac:二甲基乙酰胺
deac:二乙基乙酰胺
nmp:1-甲基-2-吡咯烷酮
实施例1:bpda-ppda(98.9:100)聚酰亚胺前体的制备
经受氮气流的反应器中填充100gn,n-二甲基丙酰胺(dmpa),然后在将反应器的温度保持在25℃的同时在其中溶解6.243g(57.726mmol)对苯二胺(p-pda)。在相同的温度下向p-pda溶液中添加16.797g(57.091mmol)3,3',4,4'-联苯羧酸二酐(s-bpda)和56.96gdmpa,通过搅拌预定的一段时间使其溶解以获得聚酰亚胺前体。
将以上有机溶剂添加至以上获得的聚酰亚胺前体中,使得其固体含量为12.8重量%,从而制备聚酰亚胺前体溶液。
实施例2:bpda-ppda-pa(98.9:100:2.2)聚酰亚胺前体的制备
经受氮气流的反应器中填充100gdmpa,然后在将反应器的温度保持在25℃的同时在其中溶解6.192g(57.259mmol)对苯二胺(p-pda)。在相同的温度下向p-pda溶液中添加16.661g(56.629mmol)3,3',4,4'-联苯羧酸二酐(s-bpda)和56.96gdmpa,通过搅拌规定的时间使其溶解以使聚酰胺酸聚合。向其中添加0.187g(1.260mmol)邻苯二甲酸酐(pa)并搅拌预定的时间以获得聚酰亚胺前体。
将以上有机溶剂添加至以上获得的聚酰亚胺前体中,使得其固体含量为12.8重量%,从而制备聚酰亚胺前体溶液。
实施例3:bpda-ppda-tfmb(98.9:95:5)聚酰亚胺前体的制备
经受氮气流的反应器中填充100gdmpa,然后在将反应器的温度保持在25℃的同时在其中溶解5.777g(53.421mmol)对苯二胺(p-pda)和0.900g(2.812mmol)双(三氟甲基)联苯胺(tfmb)。在相同的温度下向p-pda和tfmb的溶液中添加16.363g(55.614mmol)3,3',4,4'-联苯羧酸二酐(s-bpda)和56.96gdmpa,通过搅拌规定的时间使其溶解以使聚酰胺酸聚合。在搅拌预定的时间之后,获得聚酰亚胺前体。
将以上有机溶剂添加至以上获得的聚酰亚胺前体中,使得其固体含量为12.8重量%,从而制备聚酰亚胺前体溶液。
实施例4:bpda-ppda-tfmb(98.9:90:10)聚酰亚胺前体的制备
经受氮气流的反应器中填充100gdmpa,然后在将反应器的温度保持在25℃的同时在其中溶解5.335g(49.332mmol)对苯二胺(p-pda)和1.775g(5.481mmol)双(三氟甲基)联苯胺(tfmb)。在相同的温度下向p-pda和tfmb的溶液中添加15.950g(54.221mmol)3,3',4,4'-联苯羧酸二酐(s-bpda)和56.96gdmpa,通过搅拌规定的时间使其溶解以使聚酰胺酸聚合。在搅拌预定的时间之后,获得聚酰亚胺前体。
将以上有机溶剂添加至以上获得的聚酰亚胺前体中,使得其固体含量为12.8重量%,从而制备聚酰亚胺前体溶液。
实施例5:s-bpda-ppda-tfmb/pa(98.9:95:5:2.2)聚酰亚胺前体的制备
经受氮气流的反应器中填充100gdmpa,然后在将反应器的温度保持在25℃的同时在其中溶解5.731g(52.999mmol)对苯二胺(p-pda)和0.893g(2.789mmol)双(三氟甲基)联苯胺(tfmb)。在相同的温度下向p-pda和tfmb的溶液中添加16.234g(55.175mmol)3,3',4,4'-联苯羧酸二酐(s-bpda)和56.96gdmpa,通过搅拌规定的时间使其溶解以使聚酰胺酸聚合。向其中添加0.182g(1.227mmol)邻苯二甲酸酐(pa)并搅拌预定的时间以获得聚酰亚胺前体。
将以上有机溶剂添加至以上获得的聚酰亚胺前体中,使得其固体含量为12.8重量%,从而制备聚酰亚胺前体溶液。
实施例6:s-bpda-ppda-tfmb/pa(98.9:90:10:2.2)聚酰亚胺前体的制备
经受氮气流的反应器中填充100gdmpa,然后在将反应器的温度保持在25℃的同时在其中溶解5.294g(48.953mmol)对苯二胺(p-pda)和1.742g(5.439mmol)双(三氟甲基)联苯胺(tfmb)。在相同的温度下向p-pda和tfmb的溶液中添加15.827g(53.794mmol)3,3',4,4'-联苯羧酸二酐(s-bpda)和56.96gdmpa,通过搅拌规定的时间使其溶解以使聚酰胺酸聚合。向其中添加0.177g(1.197mmol)邻苯二甲酸酐(pa)并搅拌预定的时间以获得聚酰亚胺前体。
将以上有机溶剂添加至以上获得的聚酰亚胺前体中,使得其固体含量为12.8重量%,从而制备聚酰亚胺前体溶液。
比较例1:s-bpda-ppda(98.9:100)聚酰亚胺前体的制备
经受氮气流的反应器中填充100gn-甲基-2-吡咯烷酮(nmp),然后在将反应器的温度保持在25℃的同时在其中溶解6.243g(57.726mmol)对苯二胺(p-pda)。在相同的温度下向p-pda溶液中添加16.797g(57.091mmol)3,3',4,4'-联苯羧酸二酐(s-bpda)和56.96gnmp,通过搅拌预定的时间使其溶解以获得聚酰亚胺前体。
将以上有机溶剂添加至以上获得的聚酰亚胺前体中,使得其固体含量为12.8重量%,从而制备聚酰亚胺前体溶液。
比较例2:s-bpda-ppda-tfmb(98.9:95:5)聚酰亚胺前体的制备
经受氮气流的反应器中填充100gn-甲基-2-吡咯烷酮(nmp),然后在将反应器的温度保持在25℃的同时在其中溶解5.777g(53.421mmol)对苯二胺(p-pda)和0.900g(2.812mmol)双(三氟甲基)联苯胺(tfmb)。在相同的温度下向p-pda和tfmb的溶液中添加16.363g(55.614mmol)3,3',4,4'-联苯羧酸二酐(s-bpda)和56.96gnmp,通过搅拌预定的时间使其溶解以使聚酰胺酸聚合。在搅拌规定的时间之后,获得聚酰亚胺前体。
将以上有机溶剂添加至以上获得的聚酰亚胺前体中,使得其固体含量为12.8重量%,从而制备聚酰亚胺前体溶液。
比较例3:s-bpda-ppda-tfmb(98.9:90:10)聚酰亚胺前体的制备
经受氮气流的反应器中填充100gn-甲基-2-吡咯烷酮(nmp),然后在将反应器的温度保持在25℃的同时在其中溶解5.335g(49.332mmol)对苯二胺(p-pda)和1.775g(5.481mmol)双(三氟甲基)联苯胺(tfmb)。在相同的温度下向p-pda和tfmb的溶液中添加15.950g(54.221mmol)3,3',4,4'-联苯羧酸二酐(s-bpda)和56.96gnmp,通过搅拌规定的时间使其溶解以使聚酰胺酸聚合。在搅拌预定的时间之后,获得聚酰亚胺前体。
将以上有机溶剂添加至以上获得的聚酰亚胺前体中,使得其固体含量为12.8重量%,从而制备聚酰亚胺前体溶液。
比较例4:s-bpda-ppda-tfmb/pa(98.9:95:5:2.2)聚酰亚胺前体的制备
经受氮气流的反应器中填充100gn-甲基-2-吡咯烷酮(nmp),然后在将反应器的温度保持在25℃的同时在其中溶解5.731g(52.999mmol)对苯二胺(p-pda)和0.893g(2.789mmol)双(三氟甲基)联苯胺(tfmb)。在相同的温度下向p-pda和tfmb的溶液中添加16.234g(55.175mmol)3,3',4,4'-联苯羧酸二酐(s-bpda)和56.96gnmp,通过搅拌规定的时间使其溶解以使聚酰胺酸聚合。向其中添加0.182g(1.227mmol)邻苯二甲酸酐(pa)并搅拌预定的时间以获得聚酰亚胺前体。
将以上有机溶剂添加至以上获得的聚酰亚胺前体中,使得其固体含量为12.8重量%,从而制备聚酰亚胺前体溶液。
比较例5:s-bpda-ppda-tfmb/pa(98.9:90:10:2.2)聚酰亚胺前体的制备
经受氮气流的反应器中填充100gn-甲基-2-吡咯烷酮(nmp),然后在将反应器的温度保持在25℃的同时在其中溶解5.294g(48.953mmol)对苯二胺(p-pda)和1.742g(5.439mmol)双(三氟甲基)联苯胺(tfmb)。在相同的温度下向p-pda和tfmb的溶液中添加15.827g(53.794mmol)3,3',4,4'-联苯羧酸二酐(s-bpda)和56.96gnmp,通过搅拌预定的时间使其溶解以使聚酰胺酸聚合。向其中添加0.177g(1.197mmol)邻苯二甲酸酐(pa)并搅拌规定的时间以获得聚酰亚胺前体。
将以上有机溶剂添加至以上获得的聚酰亚胺前体中,使得其固体含量为12.8重量%,从而制备聚酰亚胺前体溶液。
参照例1:s-bpda-ppda-oda(98.9:95:5)聚酰亚胺前体的制备
经受氮气流的反应器中填充100gdmpa,在将反应器的温度保持在25℃的同时在其中溶解5.863g(54.215mmol)对苯二胺(p-pda)和0.571g(2.853mmol)4,4'-氧二苯胺(oda)。在相同的温度下向p-pda和oda的溶液中添加16.606g(56.440mmol)3,3',4,4'-联苯羧酸二酐(s-bpda)和56.96gnmp,通过搅拌规定的时间使其溶解以获得聚酰亚胺前体。
将以上有机溶剂添加至以上获得的聚酰亚胺前体中,使得其固体含量为12.8重量%,从而制备聚酰亚胺前体溶液。
参照例2:s-bpda-ppda-oda(98.9:95:5)聚酰亚胺前体的制备
经受氮气流的反应器中填充100gdmpa,在将反应器的温度保持在25℃的同时在其中溶解5.843g(54.034mmol)对苯二胺(p-pda)和0.646g(2.844mmol)4,4'-二氨基苯甲酰苯胺(4,4'-daba)。在相同的温度下向p-pda和4,4'-daba的溶液中添加16.550g(56.252mmol)3,3',4,4'-联苯羧酸二酐(s-bpda)和56.96gnmp,通过搅拌预定的时间使其溶解以获得聚酰亚胺前体。
将以上有机溶剂添加至以上获得的聚酰亚胺前体中,使得其固体含量为12.8重量%,从而制备聚酰亚胺前体溶液。
实验例1
通过以下方法测量实施例和比较例中制备的每种聚酰亚胺前体溶液的动态残余应力和cte,结果示于表2中。
1)残余应力
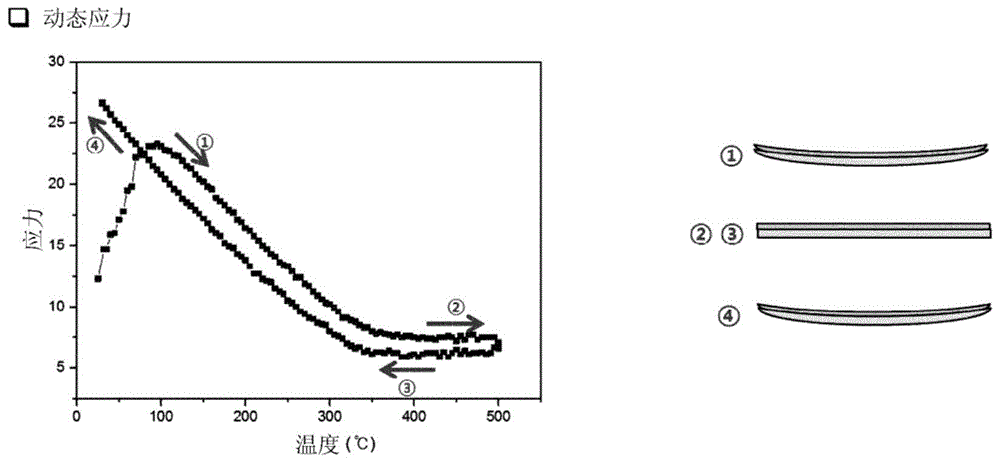
将聚酰亚胺前体溶液以6μm的厚度旋涂到厚度为525μm±45μm的4英寸硅晶片上,其中预先测量硅晶片的弯曲度。通过在残余应力测量装置(型号flx-2320,kla-tencorcorp.)的热板中在100℃至500℃的温度范围内以6℃/分钟的速率加热来使涂覆有聚酰亚胺的晶片固化,然后冷却并使用残余应力测量装置测量涂覆有聚酰亚胺的晶片的弯曲度以评估温度升高时的残余应力。结果示于表2中。此外,图1和图2示出了对于使用实施例1和比较例1的聚酰亚胺前体溶液制备的样品测量的残余应力变化的图。
2)热膨胀系数
将聚酰亚胺前体溶液旋涂在玻璃基底上。使涂覆有聚酰亚胺前体溶液的玻璃基底在烘箱中以6℃/分钟的速率经受加热,并在120℃下保持10分钟,在460℃下保持55分钟以进行固化。在固化之后,将玻璃基底浸入水中以将形成在玻璃基底上的膜分离。将分离的膜在设定为100℃的烘箱中干燥以制备6μm厚的聚酰亚胺膜。将膜切割成5mm×20mm的尺寸,使用附件将样品装载在taq400上。膜的实际测量长度为16mm,牵拉膜的力设定为0.02n。在100℃至460℃的温度范围内以4℃/分钟的加热速率进行第一加热过程,然后在460℃至100℃的温度范围内以4℃/分钟的冷却速率进行冷却过程。
然后,当使经冷却的样品以5℃/分钟的速率从100℃加热至460℃时,用tma测量热膨胀系数。测量的热膨胀系数示于表2中。此外,图2示出了对于使用实施例1和比较例1的聚酰亚胺前体溶液制备的膜测量的cte变化和残余应力变化的结果。
[表2]
根据表2,使用作为具有正的logp值的有机溶剂的dmpa的实施例1至6的聚酰亚胺膜在350℃至500℃的温度下表现出3或更小的应力变化,而使用作为具有负的logp值的有机溶剂的nmp的比较例1至5的聚酰亚胺膜在350℃至500℃的温度下表现出10或更大的应力变化。
同时,除了bpda-pda的骨架之外,还使用在芳族环内具有连接结构(例如,-o-或-c(o)nh-)的二胺的参照例1和2的膜在350℃至500℃的温度下表现出大于实施例的应力变化的应力变化。
图1示出了对于使用作为具有正的logp值的有机溶剂的dmpa的实施例1中制备的聚酰亚胺膜测量的动态应力关于时间的结果。根据图1的图,本发明的聚酰亚胺膜在350℃至500℃的温度下经受加热和冷却时表现出3或更小的应力变化,这不经历由于残余应力引起的应力反向。
此外,由于使用具有负的logp值的nmp的比较例1至5中制备的聚酰亚胺膜在350℃至500℃的温度下表现出负的cte,这意味着在上述温度范围下产生收缩。图2示出了对于使用实施例1和比较例1的聚酰亚胺前体溶液制备的膜测量的cte变化和残余应力变化的结果。如图2所示,使用dmpa制备的聚酰亚胺膜在350℃或更高的高温下的热处理期间几乎没有经历应力变化。相比之下,使用nmp制备的膜由于在高温下的负cte行为而经历收缩,从而发生应力反向(这改变应力的方向)并且使形成在聚酰亚胺膜上的无机层变形,从而在无机层中产生裂纹。
因此,本发明的聚酰亚胺膜在350℃至500℃的温度下具有0至3的应力变化和正的cte值,从而有效地抑制当通过350℃或更高的高温过程在聚酰亚胺膜上形成siox层时,由于应力反向而引起的裂纹的产生。例如,如图3所示,在制造tft器件时,无机层通过在350℃或更高的高温下沉积形成,并且伴随着350℃或更高的高温过程,例如脱氢过程和活化过程。此时,即使通过在350℃或更高的高温下沉积来形成无机层例如缓冲层,本发明的聚酰亚胺膜也不经历收缩或应力反向,从而减少无机层中裂纹的产生。这可以解决可能由裂纹引起的电特性的损坏,例如柔性显示器的可恢复的残像和电流减小。
虽然已经参照本发明的附图和实施方案特别示出和描述了本发明,但本领域普通技术人员应理解,本发明的范围不限于此,并且可以在其中做出各种改变和修改。因此,本发明的实际范围将由所附权利要求及其等同方案来限定。
- 还没有人留言评论。精彩留言会获得点赞!