使用核苷酸靶识别的靶序列特异性改变技术的制作方法
本发明涉及用于靶向靶核苷酸序列的方法、用于特异性改变靶核苷酸序列的方法和用于抑制靶基因表达的方法,其中利用crispr(规律成簇间隔短回文重复)i-d型系统的核苷酸靶识别,以及涉及在所述方法中使用的包含cas(crispr相关)蛋白和引导rna的复合物。
背景技术:
细菌和古细菌具有crispr系统作为针对病毒和异源外源质粒的适应性免疫系统。crispr系统使用与入侵的dna序列互补的低分子rna(称为引导rna或grna),以促进靶外源dna的靶向和降解。此时,需要与grna结合以形成复合物的cas蛋白。crispr系统包括i型、ii型、iii型和v型系统。在任何系统中,cas蛋白-grna复合物作用于靶序列,以引起病毒和外源质粒的干扰。在ii型和v型系统中,干扰机制涉及通过整合蛋白在靶蛋白上的dna双链断裂,所述整合蛋白具有保留grna结合的蛋白质结构域和ruvc样dna切割蛋白质结构域。对于iii型系统,已在体外和体内证实与ii型系统不同,干扰由通过5至8种cas蛋白和grna的复合物切割靶rna序列引起。
近年来,已开发了使用crisprii型和v型系统的基因组编辑技术,其中cas9和cpf1用作cas蛋白。cas9和cpf1需要在靶序列的附近由约2至5个核苷酸组成的序列,其被称为前间区序列邻近基序(pam)序列,以便识别靶dna。已在体外和体内证实,cas9-grna复合物和cpf1-grna复合物是序列特异性rna引导的核酸内切酶,其在pam序列附近的靶位点处引起dna双链断裂。
另一方面,关于crispri型系统,已在来自各种细菌的基因组序列中鉴定出多个亚型,并且亚型已命名为i-a、i-b、i-c、i-d、i-e、i-f和i-u型。在这些亚型中,衍生自大肠杆菌(escherichiacoli)的i-e型系统已得到最多研究,并且已证实由六种cas蛋白(cas3、cse1、cse2、cas7、cas5、cas6e)和grna组成的复合物促进靶dna序列的降解。然而,对于除了亚型(i-c型)的其它亚型,几乎没有阐明对于干扰效应所需的cas蛋白组分、grna序列、决定靶dna的pam序列等。另外,作为使用衍生自crispri型系统的cas蛋白的技术,已报道了用于抑制靶基因表达的方法,其包括使用编码衍生自crispri型系统的cas蛋白的重组核酸分子(专利文献1),以及用于改变靶核酸的方法,其包括使用衍生自crispri型系统的cas蛋白与其它蛋白的复合物(专利文献2和专利文献3)。然而,从未报道通过衍生自crispri型系统的rna引导的核酸内切酶,用于切割和改变靶dna分子的双链的技术。
引用列表
专利文献
专利文献1:wo2015/155686
专利文献2:jp-a2015-503535
专利文献3:wo2017/043573
发明概述
技术问题
在常规的crisprii型和v型系统中,待用于靶向的rna分子限于决定靶特异性的约2至5个核苷酸的pam序列之前或之后的约20个核苷酸的rna分子。因此,常规的crisprii型和v型系统具有以下问题:存在其中不能设计靶的基因座,以及可能切割相似的序列。需要开发不具有该问题的新型靶向系统和新型rna引导的核酸内切酶。
问题的解决方案
为了解决上述问题,本发明人进行深入研究。结果令人惊讶的是,从crispri-d型中发现了新型靶向系统和新型rna引导的内切核酸酶,其靶向比基因组编辑技术中常规使用的crisprii型或v型rna引导的内切核酸酶的靶序列更长的序列,然后发现该新型靶向系统和rna引导的核酸内切酶可以用于基因组编辑技术中,用于允许对靶核苷酸序列的改变。因此,完成了本发明。
即,本发明提供了:
[1]一种用于靶向靶核苷酸序列的方法,所述方法包括将以下引入细胞内:
(i)crispri-d型相关蛋白cas5d、cas6d和cas7d,或者编码该蛋白质的核酸,和
(ii)引导rna或编码引导rna的dna,所述引导rna包含与靶核苷酸序列互补的序列、以及在互补序列之前和之后的衍生自crispr基因座的共同重复序列;
[2]一种用于改变靶核苷酸序列的方法,所述方法包括将以下引入细胞内:
(i)crispri-d型相关蛋白cas3d、cas5d、cas6d、cas7d和cas10d,或者编码该蛋白质的核酸,和
(ii)引导rna或编码引导rna的dna,所述引导rna包含与靶核苷酸序列互补的序列、以及在互补序列之前和之后的衍生自crispr基因座的共同重复序列;
[3]一种用于抑制靶基因表达的方法,所述方法包括将以下引入细胞内:
(i)crispri-d型相关蛋白cas5d、cas6d和cas7d或者编码该蛋白的核酸,和
(ii)引导rna或编码引导rna的dna,所述引导rna包含与靶基因序列的至少一部分互补的序列、以及在互补序列之前和之后的衍生自crispr基因座的共同重复序列;
[4]根据[1]至[3]中任一项的方法,其中所述引导rna包含与靶核苷酸序列互补的由20至50个核苷酸组成的序列;
[5]根据[2]或[4]的方法,其进一步包括将供体多核苷酸引入细胞内;
[6]根据[2]、[4]和[5]中任一项的方法,其中所述改变是核苷酸缺失、插入或取代;
[7]根据[1]至[6]中任一项的方法,其中所述cas5d将5'-gth-3'(h=a、c或t)识别为前间区序列邻近基序(pam)序列;
[8]一种复合物,其包含:
(i)crispri-d型相关蛋白cas5d、cas6d和cas7d,和
(ii)引导rna,其包含与靶核苷酸序列互补的序列、以及在互补序列之前和之后的衍生自crispr基因座的共同重复序列;
[9]根据[8]的复合物,其进一步包含cas3d和cas10d;
[10]根据[8]或[9]的复合物,其中所述引导rna包含与靶核苷酸序列互补的由20至50个核苷酸组成的序列;
[11]一种表达载体,其包含:
(i)编码crispri-d型相关蛋白cas5d、cas6d和cas7d的核酸,和
(ii)编码引导rna的dna,所述引导rna包含与靶核苷酸序列互补的序列、以及在互补序列之前和之后的衍生自crispr基因座的共同重复序列;
[12]根据[11]的表达载体,其进一步包含编码cas3d和cas10d的核酸;
[13]一种dna分子,其编码根据[8]至[10]中任一项的复合物;
[14]以下用于靶向靶核苷酸序列的用途
(i)crispri-d型相关蛋白cas5d、cas6d和cas7d,或者编码该蛋白质的核酸,和
(ii)引导rna或编码引导rna的dna,所述引导rna包含与靶核苷酸序列互补的序列、以及在互补序列之前和之后的衍生自crispr基因座的共同重复序列;
[15]以下用于改变靶核苷酸序列的用途
(i)crispri-d型相关蛋白cas3d、cas5d、cas6d、cas7d和cas10d,或者编码该蛋白质的核酸,和
(ii)引导rna或编码引导rna的dna,所述引导rna包含与靶核苷酸序列互补的序列、以及在互补序列之前和之后的衍生自crispr基因座的共同重复序列;
[16]以下用于抑制靶基因表达的用途
(i)crispri-d型相关蛋白cas5d、cas6d和cas7d或者编码该蛋白质的核酸,和
(ii)引导rna或编码引导rna的dna,所述引导rna包含与靶基因序列的至少一部分互补的序列、以及在互补序列之前和之后的衍生自crispr基因座的共同重复序列;
[17]根据[14]至[16]中任一项的用途,其中所述引导rna包含与靶核苷酸序列互补的由20至50个核苷酸组成的序列;
[18]根据[15]或[17]的用途,其中所述改变是核苷酸缺失、插入或取代;
[19]根据[14]至[18]中任一项的用途,其中所述cas5d将5'-gth-3'(h=a、c或t)识别为前间区序列邻近基序(pam)序列;
[20]包含以下的复合物用于靶向靶核苷酸序列的用途:
(i)crispri-d型相关蛋白cas5d、cas6d和cas7d,和
(ii)引导rna,其包含与靶核苷酸序列互补的序列、以及在互补序列之前和之后的衍生自crispr基因座的共同重复序列;
[21]包含以下的复合物用于改变靶核苷酸序列的用途:
(i)crispri-d型相关蛋白cas3d、cas5d、cas6d、cas7d和cas10d,和
(ii)引导rna,其包含与靶核苷酸序列互补的序列、以及在互补序列之前和之后的衍生自crispr基因座的共同重复序列;
[22]包含以下的复合物用于抑制靶基因表达的用途:
(i)crispri-d型相关蛋白cas5d、cas6d和cas7d,和
(ii)引导rna,其包含与靶基因序列的至少一部分互补的序列、以及在互补序列之前和之后的衍生自crispr基因座的共同重复序列;和
[23]根据[20]至[22]中任一项的用途,其中所述引导rna包含与靶核苷酸序列互补的由20至50个核苷酸组成的序列。
发明效果
crispri-d型(下文也称为“tid”)系统的pam序列与crisprii型系统和v型系统的pam序列不同。因此,根据本发明,crispri-d型的cas蛋白的使用使得能够靶向基因座,所述基因座无法通过使用crisprii型或v型rna引导的核酸内切酶的常规基因组编辑技术靶向。此外,本发明的crispri-d型衍生的rna引导的核酸内切酶的pam序列比crisprii型和v型的pam序列更频繁地在一些生物的基因组序列上发现。因此,根据本发明,能够靶向比利用crisprii型和v型系统的常规基因组编辑技术更大数目的基因序列。此外,本发明人发现,crispri-d型系统中的grna可以靶向长度为30个核苷酸或更长的靶序列。另一方面,crisprii型或v型系统中的grna可以靶向长度为约20个核苷酸的序列。因此,本发明的crispri-d型系统显示比常规技术更稳定的结合特性和靶特异性。
因此,根据本发明,能够实现突变等位基因的生成,通过转录激活和失活的基因表达控制,以及在不能通过常规技术靶向的基因区域上通过dna修饰/组蛋白修饰蛋白质结构域的靶向的表观基因组改变。
附图简述
图1概述了本发明的crispri-d型系统的组分,以及对靶序列的靶向和切割模式。
图2显示了用于大肠杆菌基因组编辑的tid表达载体:a)pectid2质粒的结构;b)pectid3质粒的结构,pro:j23108合成启动子,t1:终止子序列stop767,rbs:核糖体结合序列,t2:终止子序列stop768(1),t3:终止子序列top768(2),t7:t7终止子序列,7d:衍生自铜绿微囊藻(microcystisaeruginosa)(下文缩写为“ma”)的cas7d,6d:macas6d,5d:macas5d,3d:macas3d,10d:macas10d,t7pro:t7启动子,crrna:tid衍生的crispr重复序列,cm:氯霉素抗性基因,p15aori:p15a质粒衍生的复制起点。
图3显示了pmw_ccdb和pmw_ccdb-pam文库质粒的结构:a)pmw_ccdb的结构,t2:rrnb2终止子序列,t1:rrnb1终止子序列,pam:前间区序列邻近基序序列,t7pro:t7启动子,ccdb:ccdb基因,km:卡那霉素抗性基因,psc101ori:psc101质粒衍生的复制起点;b)pmw_ccdb-pam质粒文库的靶序列,其中将4个随机核苷酸插入nnnn位点内,以获得pam序列筛选文库质粒,带框区域指示t7启动子,有下划线的序列指示tid靶序列,并且大写字母指示ccdb基因座。
图4显示了用于植物基因组编辑的tid表达载体:a)pegptid1质粒的结构;b)用于植物的crrna表达盒的结构;c)pegptid2质粒的结构,rb:右边界序列,lb:左边界序列,2x35s:2x花椰菜花叶病毒35s基因启动子和翻译增强子ω序列,3d:具有编码2xnls(核定位信号)的序列的macas3d,10d:具有2xnls的macas10d,7d:具有2xnls的macas7d,6d:具有2xnls的macas6d,5d:具有2xnls的macas5d,2a(1)-(4):自切割肽2a序列(1)-(4),ter:拟南芥属热休克蛋白18.2kda基因终止子,km:卡那霉素抗性基因表达盒,u6-26:拟南芥属u6snrna-26基因启动子,crrna:tid基因座衍生的crispr重复序列。
图5-1显示了使用pegptid2-pds的烟草pds基因的诱变:a)烟草pds基因上的靶序列,其中靶序列1选自第三个外显子,且靶序列2选自第六个外显子,下图中所示的靶序列中的带框部分指示pam序列,且有下划线的部分指示靶序列;b)通过农杆菌渗入法引入pegptid2-pds和gfp表达二元载体,其中携带pegptid2-pds(1)或pegptid-pds(2)的农杆菌和携带gfp表达二元质粒的农杆菌通过农杆菌渗入法被感染,并且切下其中观察到gfp表达的叶盘,并用于pds突变引入的分析。
图5-2显示了使用pegptid2-pds的烟草pds基因的定点诱变:c)通过cel-1测定分析pds突变引入,其中基因组dna由其中在图5-b)中观察到gfp表达的叶盘制备,并且通过cel-1测定分析突变的存在或不存在。三角形标记物指示通过cel-1核酸酶切割的突变的pds基因片段。
图6显示了使用pegptid2-iaa9的番茄iaa9基因诱变:a)番茄iaa9基因上的靶序列,其中靶序列1选自第二个外显子,下图中所示的靶序列中的带框部分指示pam序列,并且有下划线的部分指示靶序列;b)通过农杆菌属(agrobacterium)方法将pegptid2-iaa9引入番茄叶盘内,以获得转化的愈伤组织细胞;c)通过pcr-rflp的突变分析,其中通过pcr从基因组dna中扩增含有iaa9靶序列的区域,所述基因组dna由pegptid2-iaa9引入其内的转化的愈伤组织细胞制备,并且通过使用acci的pcr-rflp执行突变分析。空心三角形指示野生型衍生的acci切割片段,并且空心三角形上方的三角形指示未经历acci切割的突变片段。
图7显示了通过在引入pectid2-iaa9的愈伤组织中的测序的突变分析。上部序列显示了野生型iaa9序列,并且加下划线的部分指示靶序列。带框序列指示pam序列。其中发生突变的位点由插入符号或连字符显示。连字符指示核苷酸缺失。
图8显示了在引入pectid2-iaa9的再生植物中的突变分析:a)通过pcr-rflp的突变分析,其中空心三角形指示野生型衍生的acci切割片段,并且空心三角形上方的三角形指示未经历acci切割的突变片段;以及b)引入突变的番茄植物的照片,其显示了由于iaa9基因破坏的真叶形态异常。
图9显示了使用hek293细胞系的基因组编辑的实验方案。
图10显示了通过异源双链体迁移率分析的突变分析的结果。从细胞的基因组中检测到被认为衍生自突变序列的片段(黑键符号),在所述细胞内引入含有emx1基因的靶1的序列和tid基因的crrna。
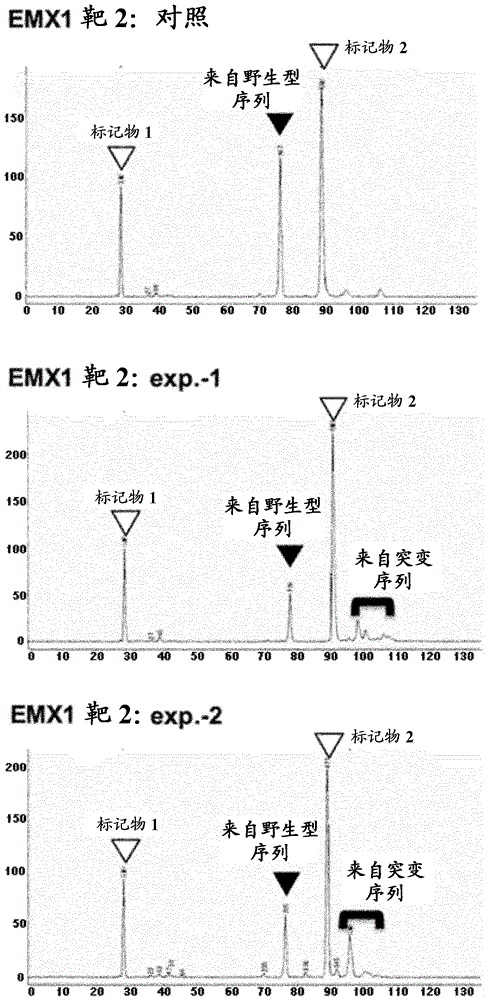
图11显示了通过异源双链体迁移率分析的突变分析的结果。从细胞的基因组中检测到被认为衍生自突变序列的片段(黑键符号),在所述细胞内引入含有emx1基因的靶2的序列和tid基因的crrna。
图12显示了突变序列的测序分析。黑色背景中的白色字母指示由tid识别的pam(前间区序列邻近序列)。带框序列指示靶序列。连字符(-)指示核苷酸缺失。黑色粗体小写字母字符指示核苷酸插入。在每个序列的右侧,显示了体细胞突变效率(其中观察到突变序列的克隆数目/分析的克隆总数目)。
图13显示了突变序列的测序分析。黑色背景中的白色字母指示由tid识别的pam(前间区序列邻近序列)。带框序列指示靶序列。连字符(-)指示核苷酸缺失。黑色粗体小写字母字符指示核苷酸插入。在每个序列的右侧,显示了体细胞突变效率(其中观察到突变序列的克隆数目/分析的克隆总数目)。
用于实施本发明的方式
本发明提供了利用crispri-d型系统的基因组编辑技术。具体地,在crispri-d型cas蛋白中,在本发明中使用cas3d、cas5d、cas6d、cas7d和cas10d。在本发明中,发现crispri-d型系统包括包含cas5d、cas6d和cas7d的靶识别模块,以及包含cas3d和cas10d的多核苷酸切割模块。
具体地,本发明的作用原理如下。
包含以下的复合物:
1)对于靶核苷酸序列的靶向(下文也称为“靶向”)是必需的grna,其包含与靶核苷酸序列互补的序列和存在于crispri-d型基因座中的共同重复序列,
2)cas5d,其识别存在于靶核苷酸序列附近的pam序列,
3)cas7d,其与1)的grna结合,并且对于靶核苷酸序列的靶向是必需的,和
4)cas6d,其执行1)的grna的加工,以及
5)包含cas10d和cas3d的复合物,所述cas10d与包含1)至4)的复合物相互作用并执行靶核苷酸序列的重塑,所述cas3d执行多核苷酸的降解,
被提供给细胞,并且在该细胞中执行
6)通过包含1)至4)的复合物靶向靶核苷酸序列,即,
7)通过包含成熟grna以及2)和3)的复合物靶向靶核苷酸序列,所述成熟grna通过经由4)的cas6d加工1)的grna获得,
并且
8)通过5)的复合物切割靶核苷酸序列上的多核苷酸。
因此,本发明提供了用于靶向靶核苷酸序列的方法(下文也称为“本发明的靶序列靶向方法”)、用于改变靶核苷酸序列的方法(下文称为“本发明的靶序列改变方法”)、以及用于抑制靶基因表达的方法(下文也称为“本发明的靶基因表达抑制方法”),其中crispri-d型系统用于方法中。此外,本发明提供了包含crispri-d型相关的cas蛋白和grna的复合物(下文也称为“本发明的复合物”)、以及包含编码复合物的核酸分子的载体,其在本发明的上述方法中使用。
(1)细胞
在本发明中,细胞可以是原核细胞或真核细胞,并无特别限制。细胞的实例包括细菌、古细菌、酵母、植物细胞、昆虫细胞和动物细胞(例如人细胞、非人细胞、非哺乳动物脊椎动物细胞、无脊椎动物细胞等)。
(2)rna引导的核酸内切酶和cas蛋白
在本发明中,“rna引导的核酸内切酶”意指包含至少一个核酸酶结构域以及与grna结合的至少一个结构域的核酸内切酶,其被grna引导至靶核苷酸序列(或靶核苷酸位点)。用于本发明中的rna引导的核酸内切酶是衍生自crispri-d型的rna引导的核酸内切酶,并且包含crispri-d型相关蛋白cas3d、cas5d、cas6d、cas7d和cas10d。在本发明中,发现cas5d、cas6d和cas7d构成促成靶识别的“靶识别模块”,并且cas3d和cas10d构成促成多核苷酸的切割的“多核苷酸切割模块”。具体地,用于本发明中的rna引导的核酸内切酶包括包含cas5d、cas6d和cas7d的靶识别模块、以及包含cas3d和cas10d的多核苷酸切割模块。
本发明中使用的cas3d、cas5d、cas6d、cas7d和cas10d可以衍生自任何细菌或古细菌。细菌和古细菌的实例包括铜绿微囊藻、阿拉伯糖醋盐杆菌(acetohalobiumarabaticum)、ammonifexdegensii、柱胞鱼腥藻(anabaenacylindrica)、多变鱼腥藻(anabaenavariabilis)、产乳酸乙酸热解纤维素菌(caldicellulosiruptorlactoaceticus)、caldilineaaerophila、clostridiumalgicarnis、crinaliumepipsammum、蓝丝菌属物种(cyanothecesp.)、静水筒孢藻(cylindrospermumstagnale)、haloquadratumwalsbyi、湖渊盐红菌(halorubrumlacusprofundi)、methanocaldococcusvulcanius、亨氏甲烷螺菌(methanospirillumhungatei)、亚洲嗜盐无色菌(natrialbaasiatica)、natronomonaspharaonis、点形念珠藻(nostocpunctiforme)、phormidesmispriestleyi、尖细颤藻(oscillatoriaacuminata)、picrophilustorridus、嗜热螺旋体(spirochaetathermophila)、stanieriacyanosphaera、酸热硫化叶菌(sulfolobusacidocaldarius)、冰岛硫化叶菌(sulfolobusislandicus)、集胞藻属物种(synechocystissp.)、thermacetogeniumphaeum、thermofilumpendens等。cas蛋白的氨基酸序列和核苷酸序列信息可从公共数据库,例如ncbigenbank获得。另外,还可以通过使用blast程序,从通过宏基因组分析等等获得的微生物基因组数据中获得来自新微生物物种的序列。在基于氨基酸序列信息,选择用于在核酸引入其内的宿主细胞中的翻译的最佳密码子之后,可以例如通过化学合成等等来构建编码cas蛋白的核酸。宿主细胞中频繁使用的密码子的使用能够增加蛋白质的表达水平。例如,可以基于氨基酸序列信息化学合成cas蛋白,或者通过经由适当的载体将编码cas蛋白的核酸引入细胞内而在细胞中产生cas蛋白等等。cas3d、cas5d、cas6d、cas7d和cas10d的每种cas蛋白可以是突变型cas蛋白,只要它保留了如本发明的作用原理中所述的每种cas蛋白的功能。
(3)引导rna
在本发明中,引导rna(grna)是这样的分子,其与靶识别模块(cas5d、cas6d和cas7d)形成复合物,以连同这些cas蛋白一起靶向靶核苷酸序列。在本发明中,grna与靶识别模块的cas7d结合。在本发明中,grna与包含cas5d、cas6d和cas7d的复合物结合,以将复合物引导至靶核苷酸序列。例如,grna与rna引导的核酸内切酶的靶识别模块结合,以将rna引导的核酸内切酶引导至靶核苷酸序列。当靶识别模块作为融合蛋白的一部分而不是rna引导的核酸内切酶存在时,grna与靶识别模块结合,以将融合蛋白引导至靶核苷酸序列。
grna包含与靶序列互补的序列,使得可以在grna与靶核苷酸序列之间形成碱基对,以及在互补序列之前和之后(在其5'末端侧和3'末端侧处)的衍生自crispri-d型基因座的共同重复序列。grna的共同重复序列部分可能具有至少一个发夹结构。例如,置于与靶核苷酸序列互补的序列的5’末端侧处的共同重复序列部分可以具有发夹结构,并且置于与靶核苷酸序列互补的序列的3’末端侧处的共同重复序列部分可以是单链的。在本发明中,grna优选具有发夹结构。
通过使用串联重复搜索程序,可以从与i-d型基因群邻近的grna基因序列区域中找到衍生自crispri-d型基因座的共同重复序列。grna中包含的共同重复序列的核苷酸长度并无特别限制,只要grna与靶识别模块相互作用以靶向靶核苷酸序列。例如,在与靶核苷酸序列互补的序列之前和之后的各共同重复序列可以具有约10至70个核苷酸的长度,例如30至50个核苷酸的长度。
grna可以含有由约10至70个核苷酸组成的序列,其与靶核苷酸序列互补。grna中包含的与靶核苷酸序列互补的序列优选为由20至50个核苷酸组成的序列,更优选为由25至45个核苷酸组成的序列,更优选为由30至40个核苷酸组成的序列,或甚至更优选为由32至37个核苷酸组成的序列,例如由32个核苷酸、33个核苷酸、34个核苷酸、35个核苷酸、36个核苷酸或37个核苷酸组成的序列。随着可以靶向的靶序列越长,通过grna的靶识别的序列特异性越大地增加。另外,随着可以靶向的靶序列越长,在grna与靶序列之间形成的碱基对的tm值就越高,且因此靶识别的稳定性越大地增加。由于可以通过关于常规基因组编辑技术中使用的rna引导的核酸内切酶(例如cas9和cpf1)的grna靶向的序列长度为约20至24个核苷酸的长度,因此与常规方法相比,本发明在序列特异性和稳定性方面是极佳的。
(4)靶核苷酸序列
在本发明中,靶核苷酸序列(如本文使用的,也称为“靶序列”)是任何核酸序列,并无特别限制,只要它是位于前间区序列邻近基序(pam)附近的序列。靶核苷酸序列可以是双链dna序列、单链dna序列或rna序列。dna的实例包括真核核基因组dna、线粒体dna、质体dna、原核基因组dna、噬菌体dna和质粒dna。在本发明中,靶核苷酸序列优选是基因组上的双链dna。如本文使用的,短语“在……附近”既包括与位置邻接又包括与位置接近。如本文使用的,“附近”既包括邻接又包括邻近。
用于crispr系统的靶识别的pam序列取决于crispr系统的类型而变。在本发明中,发现由crispri-d型系统利用的pam序列是5'-gth-3'(h=a、c或t)(实施例1)。优选地,选择位于pam序列的3'下游侧附近的序列作为靶核苷酸序列。例如,靶核苷酸序列可以是位于pam序列附近,并且存在于靶基因的内含子、编码区、非编码区或控制区中的序列。靶基因可以是任何基因并且任选地选择。
关于常规基因组编辑技术中使用的cas9和cpf1的pam序列分别为5'-ngg-3'(n=a、c、g或t)和5'-tttv-3'(v=a、c或g)。将关于tid的pam序列的出现频率(即crispr系统的候选靶数目)与关于高等植物的基因组序列中cas9和cpf1的pam序列的出现频率进行比较。结果,发现关于tid的pam序列的出现频率最高,并且tid具有比使用cas9和cpf1的常规基因组编辑技术更大数目的靶(表1)。
[表1]
(5)本发明的靶向靶序列的方法
本发明的靶向靶序列的方法的特征在于将靶识别模块(cas5d、cas6d和cas7d)和grna引入细胞内。具体地,本发明的靶序列靶向方法的特征在于将以下引入细胞内:(i)cas5d、cas6d和cas7d,或者编码这些蛋白质的核酸,以及(ii)grna或编码grna的dna。本发明的靶序列靶向方法可以在体外或体内执行。
在本发明的靶向靶序列的方法中,可以将靶识别模块作为包含cas5d、cas6d和cas7d的分离的复合物引入细胞内,或者可以将cas5d、cas6d和cas7d各自作为分离的单一蛋白质引入细胞内。在本发明的靶序列靶向方法中,靶识别模块也可以作为编码cas蛋白cas5d、cas6d和cas7d的核酸引入细胞内。核酸的实例包括rna例如mrna和dna。
编码cas蛋白的dna可以包含在例如载体中。dna序列优选可操作地连接至调节序列,例如启动子或终止子。当靶识别模块引入其内的细胞是真核细胞时,优选将核定位信号序列加入编码cas蛋白的dna中。编码cas蛋白cas5d、cas6d和cas7d的两种或更多种或所有dna可以包含在单一载体中,或可以包含在分开的载体中。载体的数目以及由待掺入每种载体内的dna编码的cas蛋白的种类和组合并无限制。当在单一载体中包含编码cas蛋白的两种或更多种dna时,dna序列可以例如经由编码自切割肽的序列彼此连接,以便被多顺反子表达。编码cas蛋白的两种或更多种dna可以以任何次序连接。
可以将grna作为rna或编码grna的dna引入细胞内。编码grna的dna可以包含在例如载体中。dna序列优选可操作地连接至调节序列,例如启动子或终止子。
编码cas蛋白的dna和编码grna的dna可以包含在同一载体中,或可以包含在分开的载体中。例如,编码cas5d、cas6d和cas7d的一种或多种或所有dna以及编码grna的dna可以包含在单一载体中。
调节序列例如启动子或终止子和核定位信号序列是本领域已知的,并且可以取决于其中靶识别模块和grna引入其内的细胞衍生自其的生物物种适当地选择。用于引入的载体可以取决于其中载体引入其内的细胞衍生自其的生物物种适当地选择,并无特别限制。载体的实例包括质粒载体、病毒载体、噬菌粒、粘粒、人工/微型染色体和转座子。
可以通过本领域已知的各种手段将靶识别模块和grna引入细胞内。此类手段的实例包括转染,例如磷酸钙介导的转染、电穿孔、脂质体转染等,病毒转导,脂转染,基因枪,显微注射,农杆菌属方法,农杆菌渗入法和peg-钙法。
靶识别模块和grna可以同时或序贯地引入细胞内。构成靶识别模块的cas5d、cas6d和cas7d,或者编码这些cas蛋白的核酸可以同时或序贯地引入细胞内。例如,在体外或体内合成的cas蛋白cas5d、cas6d和cas7d,以及在体外或体内合成的grna,可以在体外进行温育以形成复合物,并且可以将该复合物引入细胞内。
在引入靶识别模块和grna后,在合适的条件下培养细胞用于靶核苷酸序列的靶向。然后在合适的条件下培养细胞用于细胞生长和维持。培养条件可以适合于其中靶识别模块和grna引入其内的细胞衍生自其的生物物种,并且可以由本领域技术人员例如基于已知的细胞培养技术适当地确定。
根据本发明的靶向靶序列的方法,grna与靶识别模块的cas7d结合,以形成靶识别模块和grna的复合物。同时,grna与靶核苷酸序列形成碱基对。靶识别模块通过识别存在于靶核苷酸序列附近的pam序列,以序列特异性的方式靶向靶核苷酸序列。在本发明的靶序列靶向方法中,可以将cas10d进一步引入细胞内。
(6)本发明的靶向靶序列的方法
本发明的靶序列改变方法的特征在于将rna引导的核酸内切酶和grna引入细胞内。具体地,本发明的靶序列改变方法的特征在于将以下引入细胞内:(i)cas3d、cas5d、cas6d、cas7d和cas10d,或者编码该蛋白质的核酸,以及(ii)grna或编码grna的dna。本发明的靶序列改变方法包括用多核苷酸切割模块切割由本发明的靶序列靶向方法靶向的核苷酸序列。本发明的靶序列改变方法可以在体外或体内执行。在本发明中,改变包括一个或多个核苷酸的缺失、插入和取代,及其组合。
在本发明的改变靶序列的方法中,除rna引导的核酸内切酶和grna之外,还可以将供体多核苷酸引入细胞内。供体多核苷酸包含至少一个供体序列,其含有期望引入靶位点内的改变。除供体序列之外,供体多核苷酸还可以包含在供体序列的两个末端处,与靶序列的上游和下游序列具有高度同源性的序列(优选与靶序列的上游和下游序列基本上相同的序列)。供体多核苷酸可以是单链或双链dna。供体多核苷酸可以由本领域技术人员基于本领域已知的技术适当地设计。
当在本发明的改变靶序列的方法中不存在供体多核苷酸时,可以通过非同源末端连接(nhej)来修复靶核苷酸序列中的切割。已知nhej是易错的,并且在切割修复期间可能发生一个或多个核苷酸的缺失、插入或取代,或其组合。因此,可以在靶序列位点处改变序列,并且从而诱导移码或不成熟的终止密码子,以失活或敲除由靶序列区域编码的基因的表达。
当供体多核苷酸存在于本发明的改变靶序列的方法中时,将供体多核苷酸的供体序列插入靶序列位点内,或通过切割的靶核苷酸序列的同源重组修复(hdr)替换靶序列位点。结果,将期望的改变引入靶序列位点内。
可以将rna引导的核酸内切酶作为包含cas5d、cas6d、cas7d、cas3d和cas10d的分离的复合物引入细胞内,或者可以将cas5d、cas6d、cas7d、cas3d和cas10d各自作为分离的单一蛋白质引入细胞内。rna引导的核酸内切酶也可以作为编码cas蛋白cas5d、cas6d、cas7d、cas3d和cas10d的核酸引入细胞内。核酸的实例包括rna例如mrna和dna。
编码cas蛋白的dna可以包含在例如载体中,并且dna序列优选可操作地连接至调节序列,例如启动子或终止子。当rna引导的核酸内切酶引入其内的细胞是真核细胞时,优选将核定位信号序列加入编码cas蛋白的dna中。编码cas蛋白cas3d、cas5d、cas6d、cas7d和cas10d的两种或更多种或所有dna可以包含在单一载体中,或可以包含在分开的载体中。载体的数目以及由待掺入每种载体内的dna编码的cas蛋白的种类和组合并无限制。当在单一载体中包含编码cas蛋白的两种或更多种dna时,dna序列可以例如经由编码自切割肽的序列彼此连接,以便被多顺反子表达。编码cas蛋白的两种或更多种dna可以以任何次序连接。
可以将grna作为rna或编码grna的dna引入细胞内。编码grna的dna可以包含在例如载体中。dna序列优选可操作地连接至调节序列,例如启动子或终止子。
编码cas蛋白的dna和编码grna的dna可以包含在同一载体中,或可以包含在分开的载体中。例如,编码cas3d、cas5d、cas6d、cas7d和cas10d的一种或多种或所有dna以及编码grna的dna可以包含在单一载体中。
调节序列例如启动子或终止子和核定位信号序列是本领域已知的,并且可以取决于ran引导的核酸内切酶和grna引入其内的细胞的种类适当地选择。用于引入的载体可以取决于载体引入其内的细胞的种类适当地选择,并无特别限制。载体的实例包括质粒载体、病毒载体、噬菌粒、粘粒、人工/微型染色体和转座子。
可以通过本领域已知的各种手段将rna引导的核酸内切酶、grna和供体多核苷酸引入细胞内。此类手段的实例包括转染,例如磷酸钙介导的转染、电穿孔、脂质体转染等,病毒转导,脂转染,基因枪,显微注射,农杆菌属方法,农杆菌渗入法和peg-钙法。
rna引导的核酸内切酶、grna和供体多核苷酸可以同时或序贯地引入细胞内。构成rna引导的核酸内切酶的cas3d、cas5d、cas6d、cas7d和cas10d,或者编码这些cas蛋白的核酸可以同时或序贯地引入细胞内。
在引入rna引导的核酸内切酶和grna或rna引导的核酸内切酶、grna和供体多核苷酸后,在合适的条件下培养细胞用于在靶序列位点处的切割。然后在合适的条件下培养细胞用于细胞生长和维持。培养条件可以适合于rna引导的内切核酸酶和grna或rna引导的内切核酸酶、grna和供体多核苷酸引入其内的细胞所来源的生物物种,并且可以由本领域技术人员例如基于已知的细胞培养技术适当地确定。
根据本发明的改变靶序列的方法,grna与靶核苷酸序列形成碱基对,并且同时,grna与rna引导的核酸内切酶的靶识别模块相互作用,以将rna引导的核酸内切酶引导至靶序列位点。然后,rna引导的核酸内切酶的切割模块在靶序列位点处切割该序列。当切割的序列被修复时,靶序列被改变。例如,本发明的改变靶序列的方法可以用于改变基因组上的靶核苷酸序列。通过本发明的改变靶序列的方法切割基因组上的双链dna,然后在靶位点处改变。
(7)本发明的靶基因抑制的方法
本发明的靶基因表达抑制方法的特征在于将靶识别模块(cas5d、cas6d和cas7d)和grna引入细胞内。具体地,本发明的靶基因抑制的方法的特征在于将以下引入细胞内:(i)cas5d、cas6d和cas7d,或者编码该蛋白质的核酸,以及(ii)grna或编码grna的dna。在本发明的靶基因抑制的方法中,选择靶基因序列的至少一部分作为靶核苷酸序列,并且使用含有与靶序列互补的序列的grna。本发明的靶基因抑制的方法包括:当通过本发明的靶基因抑制的方法靶向靶核苷酸序列时,通过靶识别模块和grna的复合物与靶序列的结合,抑制含有靶序列的基因的表达。本发明的靶基因抑制的方法可以在体外或体内执行。根据本发明的靶基因抑制的方法,尽管靶基因序列未被切割,但含有靶序列的基因区域的功能或基因的表达通过靶识别模块和grna的复合物与靶核苷酸序列的结合而被抑制。
靶识别模块和grna,用于将其引入细胞内的方法,在引入时和在引入后的细胞培养等等如“(5)本发明的靶向靶序列的方法”中所述。在本发明的靶基因抑制的方法中,可以将cas10d进一步引入细胞内。
(8)本发明的复合物
本发明的复合物包含crispri-d型cas蛋白和grna。本发明特别提供了包含靶识别模块和grna的复合物,以及包含rna引导的核酸内切酶和grna的复合物。更具体地,提供了包含cas5d、cas6d、cas7d和grna的复合物,以及包含cas5d、cas6d、cas7d、cas3d、cas10d和grna的复合物。另外,提供了编码复合物的dna分子。本发明的复合物可以用于本发明的改变靶序列的方法,靶基因抑制的方法和靶向靶序列的方法中。通过将包含rna引导的核酸内切酶(包含cas5d、cas6d、cas7d、cas3d和cas10d的复合物)和grna的复合物引入细胞内,以允许该复合物在细胞中起作用,可以改变细胞的基因组上的靶序列。另外,通过将包含靶识别模块(包含cas5d、cas6d和cas7d的复合物)和grna的复合物引入细胞内,以允许该复合物在细胞中起作用,可以靶向细胞中的靶序列,并且可以抑制由靶序列区域编码的基因的表达。包含靶识别模块和grna的复合物可以进一步含有cas10d。
本发明的复合物可以通过常规方法在体外或体内生产。例如,可以将编码构成rna引导的核酸内切酶或靶识别模块的cas蛋白的核酸、以及grna或编码grna的dna引入细胞内,以允许复合物在细胞中形成。
本发明的复合物的实例包括但不限于包含以下的复合物:来自铜绿微囊藻的cas5d(seqidno:1)、cas6d(seqidno:2)和cas7d(seqidno:3),以及由通过guuccaauuaaucuuaagcccuauuagggauugaaacnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnguuccaauuaaucuuaagcccuauuagggauugaaac(seqidno:6;n是构成与靶核苷酸序列互补的序列的任何核苷酸)显示的序列组成的grna;以及包含以下的复合物:来自铜绿微囊藻的cas5d(seqidno:1)、cas6d(seqidno:2)、cas7d(seqidno:3)、cas3d(seqidno:4)和cas10d(seqidno:5),以及由通过guuccaauuaaucuuaagcccuauuagggauugaaacnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnguuccaauuaaucuuaagcccuauuagggauugaaac(seqidno:6;n是构成与靶核苷酸序列互补的序列的任何核苷酸)显示的序列组成的grna。在grna序列中,n的数目可以在10至70,优选20至50,更优选25至45,再更优选30至40,且再更优选32至37的范围内变化。
(9)本发明的表达载体
本发明进一步提供了表达载体,其含有编码包含cas3d、cas5d、cas6d、cas7d和cas10d的rna引导的核酸内切酶的核酸,以及编码grna的dna,所述grna包含与靶序列互补的序列和在靶序列之前和之后的衍生自crispr基因座的共同重复序列;以及表达载体,其包含编码crispri-d型相关蛋白cas5d、cas6d和cas7d的核酸,以及编码grna的dna,所述grna包含与靶序列互补的序列和在靶序列之前和之后的衍生自crispr基因座的共同重复序列。
本发明的载体是用于将cas蛋白和grna引入细胞内的载体,如“(5)本发明的靶序列靶向方法”、“(6)本发明的靶序列改变方法”和“(7)本发明的靶基因表达抑制方法”中所述。在载体引入细胞内之后,cas蛋白和grna在细胞中表达。本发明的载体还可以是其中包含在grna中的靶序列替换为含有限制性位点的任何序列的载体。在将所需的靶核苷酸序列掺入限制性位点内之后,使用此类载体。任何序列可以是例如在crispri-d型基因座上存在的间隔区序列或间隔区序列的一部分。
(10)包含本发明的靶识别模块的融合蛋白
本发明进一步提供了包含靶识别模块和功能性多肽的融合蛋白。当将融合蛋白和grna引入细胞内时,通过靶识别模块和grna的作用,将融合蛋白引导至细胞中的靶核苷酸序列或靶基因,并且通过功能性多肽的作用改变或修饰靶核苷酸序列或靶基因。因此,本发明进一步提供了用于改变或修饰靶核苷酸序列或靶基因的方法,其包括将融合蛋白和grna引入细胞内。此外,本发明提供了包含融合蛋白和grna的复合物。
功能性多肽是显示出对靶序列的任何功能的多肽,并且是除cas3d和cas10d外的多肽。功能性多肽的实例包括但不限于限制性酶、转录因子、dna甲基化酶、组蛋白乙酰化酶、荧光蛋白;多核苷酸切割模块,例如限制性酶的核苷酸切割模块;基因表达调节模块,例如转录因子的转录激活模块和转录阻遏模块;以及表观基因组修饰模块,例如dna甲基化酶的甲基化模块和组蛋白乙酰化酶的组蛋白乙酰化模块。荧光蛋白的实例是gfp。例如,以与本发明的改变靶序列的方法相同的方式,通过将包含靶识别模块和多核苷酸切割模块的融合蛋白连同grna一起引入细胞内,可以改变靶序列。例如,通过将包含靶识别模块和基因表达调节模块或表观基因组修饰模块的融合蛋白连同grna一起引入细胞内,可以修饰靶序列以调节靶基因的表达。例如,通过将包含靶识别模块和荧光蛋白的融合蛋白连同grna一起引入细胞内,可以荧光标记靶序列的附近。
下文显示了本发明的实施例。然而,本发明并不限于所述实施例。
实施例
作为一个实施方案,克隆并随后使用了衍生自crispri-d型(下文也称为“tid”)基因座(其衍生自铜绿微囊藻)的一组基因(cas3d、cas5d、cas6d、cas7d、cas10d)。对于实施例中的dna序列的加工和构建,使用了人工基因化学合成、pcr、限制性酶处理、连接或吉布森组装法。另外,sanger法或下一代测序法用于测定核苷酸序列。
实施例1.大肠杆菌中的基因组编辑
在本实施例中,证实了本发明的技术在典型的细菌模型生物大肠杆菌中有效地起作用。
(1)tid基因表达质粒的构建
克隆了衍生自铜绿微囊藻(下文也称为“铜绿微囊藻(m.aeruginosa)”)的crispri-d型基因座(下文也称为“tid基因座”)的基因群。基于衍生自来自铜绿微囊藻的tid基因座的cas5d、cas6d、cas7d、cas3d和cas10d的氨基酸序列信息,人工化学合成了编码每种cas蛋白的大肠杆菌密码子优化的序列(seqidno:7至11)。包含在每个cas蛋白编码基因上游的j23108合成启动子(seqidno:12)或合成核糖体结合序列(seqidno:13)、以及在每个cas蛋白编码基因下游的终止子序列(seqidno:14至17)的dna片段,连接到质粒载体pacyc184(由nippongene制造)内,以构建pectid1。另外,提取存在于来自铜绿微囊藻的crispri-d型基因座附近的crispr重复序列(crrna,seqidno:18),并且合成含有在t7启动子(seqidno:19)的控制下的crispr重复序列的crrna表达盒(seqidno:20)。crrna表达盒含有大肠杆菌ccdb基因的启动子区序列,其为本实施例中的靶序列。将crrna表达盒序列掺入pectid1内,以构建pectid2(图2a)。此外,含有cas5d、cas6d和cas7d基因表达盒的pectid3被构建为tid表达质粒载体,用于基因组编辑而无需dna双链断裂(图2b)。表2中显示了本实施例中使用的启动子、终止子、crispr重复序列和crrna表达盒序列。
[表2]
(2)前间区序列邻近基序(pam)文库的构建
在本实施例中,使用了合成的ccdb基因盒(seqidno:21)(表3),其中作为靶dna,将t7启动子序列连接到大肠杆菌ccdb基因的上游。tid的靶序列是包含ccdb基因上游的t7启动子区的35个核苷酸的序列。将合成的ccdb基因盒连接至质粒载体pmw219(由nippongene制造)中的多克隆位点,以构建pmw_ccdb1(图3a)。
crispr系统识别位于靶序列附近的前间区序列邻近基序(pam)序列,并且经由grna与靶序列结合。由于本实施例中使用的铜绿微囊藻tid的pam序列是未知的,因此构建了用于测定铜绿微囊藻tid的pam序列的pam序列文库质粒。通过使用人工化学dna合成和pcr,将随机的四核苷酸序列引入pmw_ccdb1的t7启动子的上游(图3b)。将构建的pmw_ccdb-pam文库质粒引入保留ccdb抗性的ccdb抗性大肠杆菌细胞系(由thermofisherscientific制造)内,然后制备质粒。
[表3]
(3)铜绿微囊藻tid系统中的pam序列的测定
关于tid的pam序列通过使用pectid3-t7进行测定,所述pectid3-t7已在pmw_ccdb-pam文库质粒上插入与t7启动子区互补的35个核苷酸的序列。将pectid3-t7引入大肠杆菌bl21ai菌株(由thermofisherscientific制造)内,以获得用于ccdb基因基因组编辑的大肠杆菌宿主菌株。bl21ai[pectid3-t7]菌株表达对于靶序列识别所需的cas5d、cas6d和cas7d蛋白。cas5d/cas6d/cas7d-crrna复合物识别与适当的pam序列邻近的靶序列,并且与靶序列结合以抑制t7启动子(其为靶序列)的功能,尽管它不切割靶序列。
在补充阿拉伯糖的培养基中诱导通过引入bl21ai菌株内的pmw-ccdb-pam的ccdb表达,并且杀死不具有ccdb抗性的bl21ai细胞。当将pmw_ccdb-pam文库质粒引入bl21ai细胞(tid表达质粒已预先引入其内)内时,由pectid3质粒表达的cas5d/cas6d/cas7-crrna结合具有待由tid识别的适当pam序列的pmw_ccdb-pam文库质粒的t7启动子,从而抑制ccdb蛋白的产生,并且因此大肠杆菌细胞可以生长。由生长的大肠杆菌菌落制备pmw_ccdb-pam文库质粒,并且通过测序分析pam序列,以测定铜绿微囊藻tid的pam序列。
通过化学感受态细胞方法,将大量制备的pmw_ccdb-pam文库质粒引入bl21ai[pectid3-t7]菌株内。在含有25mg/l氯霉素、25mg/l卡那霉素和1%葡萄糖的lb琼脂培养基上,选择保留pmw_ccdb-pam文库质粒和pectid3-t7的bl21ai细胞。从因此获得的大肠杆菌菌落中,收集约1×107个菌落,用不含抗生素和葡萄糖的lb液体培养基洗涤几次,然后以1×106个细胞/ml悬浮于含有1%阿拉伯糖的lb液体培养基中。悬浮液在37ºc下伴随振荡培养2小时,以通过阿拉伯糖诱导在t7启动子的控制下的crrna和ccdb表达。然后,200μl悬浮液在含有25mg/l氯霉素、25mg/l卡那霉素和1%阿拉伯糖的lb琼脂培养基上进行划线培养。在37ºc下培养过夜后,收集细菌菌落。从因此收集的约500个菌落中,制备它们的质粒,并且使pam序列的附近经受测序分析。在tid表达质粒的存在下拯救的pmw_ccdb-pam文库质粒的pam序列含有序列5'-ngth-3'(n=a、c、g或t;h=a、c或t)。pam序列的使用频率对于ngta为28%,对于ngtc为33%,且对于ngtt为38%。因此,发现由tid利用的pam序列是5'-gth-3'(h=a、c或t)。
(4)大肠杆菌中的基因组编辑
构建了质粒pmw_ccdb-pamgta、pmw_ccdb-pamgtc和pmw_ccdb-pamgtt,其含有使用pectid3-t7和pmw_ccdb-pam文库质粒测定的三种类型的pam序列,并且连同pectid2-i7一起引入bl21ai菌株内。在含有25mg/l氯霉素、25mg/l卡那霉素和1%葡萄糖的lb琼脂培养基上,选择保留pmw_ccdb-pamgta/pectid2-t7、pmw_ccdb-pamgtc/pectid2-t7和pmw_ccdb-pamgtt/pectid2-t7的bl21ai细胞,然后通过测序分析发现含有引入各细菌细胞内的质粒。随后,保留正确质粒的bl21ai细胞在含有25mg/l氯霉素、25mg/l卡那霉素和1%阿拉伯糖的lb琼脂培养基上进行划线培养,然后在37ºc下培养过夜。结果,所有细菌细胞都不生长,其很可能由在cas3d和cas10d的存在下质粒dna上的双链dna断裂引起。
实施例2.高等植物中的基因组编辑
在本实施例中,作为高等真核生物中的基因组编辑的一个实施方案,证实了本发明的技术在本塞姆氏烟草(nicotianabenthamiana)和番茄(solanumlycopersicum)中有效地起作用。
(1)用于在高等植物细胞中的tid基因表达的二元载体的构建
根据拟南芥属和烟草中的频率,基于衍生自铜绿微囊藻的tid基因座的cas5d、cas6d、cas7d、cas3d和cas10d的氨基酸序列信息,人工化学合成了编码每种cas蛋白的双子叶植物密码子优化序列。制备了dna片段,其包含核定位信号序列(seqidno:22,seqidno:23),其含有在cas蛋白编码基因各自的5'上游串联排列的两个核定位信号;以及在cas蛋白编码基因之间的自切割肽2a序列(seqidno:24-28)。包含串联排列的两个花椰菜花叶病毒35s基因启动子的启动子序列(2×35s启动子;seqidno:29)和翻译增强子ω序列,与经由2a肽序列彼此融合的五个tid基因片段的5'上游连接,并且拟南芥属热休克蛋白18.2kda基因终止子序列(seqidno:30)与经由2a肽序列彼此融合的五个tid基因片段的3'下游连接,并且从而制备了tid基因表达盒。将tid基因表达盒克隆到二元质粒载体pcambia2300内,以构建pegptid1(图4a)。对于用于植物的crrna表达盒,人工化学合成了dna(seqidno:31),其中在两个crrna序列之间放置含有两个限制性酶bsai位点的间隔区序列,使得35个核苷酸的任何序列可以连接到bsai位点内。拟南芥属u6snrna-26基因启动子序列(seqidno:32)在crrna表达盒序列的5'上游处连接,而聚t序列在crrna表达盒序列的3'下游处连接(图4b)。用于植物的crrna表达盒连接到pegptidd1的rb序列和2×35s启动子之间,以构建pegptid2,其用作用于植物基因组编辑的tid基因表达二元质粒载体(图4c)。核定位序列在pegptidd1和pegptidd2内与其连接的编码每种cas蛋白的双子叶植物密码子优化序列显示于seqidnos:33至37中。表4中显示了本实施例中使用的核定位信号序列、自切割肽2a序列、启动子、终止子和crrna表达盒序列。
[表4-1]
[表4-2]
(2)本塞姆氏烟草中的基因组编辑
在烟草的实施例中,选择八氢番茄红素去饱和酶(pds)基因作为用于引入突变的靶序列(图5-1a)。从烟草pds基因的第三个外显子中选择靶序列1(靶1,seqidno:38),并且将靶1的人工化学合成的dna连接到用于植物的crrna表达盒内,以构建pegptid2-pds(1)。类似地,从第六个外显子中选择靶序列2(靶2,seqidno:39),并且将靶2的人工化学合成的dna连接到用于植物的crrna表达盒内,以构建pegptid2-pds(2)。将因此构建的二元载体引入根癌农杆菌(agrobacteriumtumefaciens)菌株gv2260内。通过农杆菌渗入法执行靶向烟草pds的tid表达载体引入烟草细胞内。保留pegptid2-pds(1)或pegptidd2-pds(2)的农杆菌属细胞和保留gfp表达二元载体的农杆菌属细胞分开进行培养,然后共感染本塞姆氏烟草的真叶(图5-1b)。在共感染后三天,基因组dna由叶盘中发出gfp荧光的区域制备,然后用作模板以pcr扩增含有靶序列的300-500bppds基因片段。pcr扩增的片段用于cel-1测定,以分析是否将突变引入pds基因内。作为对照,使用仅gfp表达二元载体引入其内的烟草叶盘。当仅引入gfp表达载体时,在pds基因上未观察到突变。相比之下,当同时引入pegptid2-pds和gfp表达载体时,在pds基因的每个靶序列上观察到突变的引入(图5-2c)。表5中显示了靶序列1和2。
[表5]
(3)番茄中的基因组编辑
在番茄的实施例中,选择aux/iaa转录因子iaa9基因作为用于引入突变的靶序列(图6a)。从番茄iaa9基因的第二个外显子中选择靶序列1(seqidno:40)(表6),并且将靶1的人工化学合成的dna连接到用于植物的crrna表达盒内,以构建pegptid2-iaa9。将构建的二元载体引入根癌农杆菌菌株gv2260内。使用来自番茄子叶的叶盘,通过农杆菌属方法,执行靶向番茄iaa9基因的tid表达载体引入番茄细胞内。在含有100mg/l卡那霉素和1.5mg/lt-玉米素的ms固化培养基上培养用农杆菌属共接种的叶盘,以获得其中发生了在pegptid2-iaa9上的t-dna区域的基因引入的愈伤组织(图6b)。关于限制性酶acci的识别序列存在于iaa9的靶序列中。当突变由于通过tid的基因组编辑而引入时,acci识别位点消失。因此,执行使用acci的pcr限制性酶长度多态性(rflp)分析,以分析在iaa9的靶序列上发生的突变。基因组dna由获得的转化的愈伤组织制备,并且用作模板以pcr扩增含有iaa9的靶序列的大约300个碱基的区域。pcr片段用acci进行消化。发现来自pegptid2-iaa9引入其内的愈伤组织培养物的pcr片段含有由于iaa9靶序列中的突变引入而未被acci消化的序列(图6c)。测定来自pegptid2-iaa9引入其内的愈伤组织的pcr片段的核苷酸序列。结果,发现1至4个核苷酸的核苷酸缺失紧在pam序列之后引入到iaa9的靶序列上(图7)。
pegptid2-iaa9引入其内的愈伤组织在含有100mg/l卡那霉素和1.0mg/lt-玉米素的ms固化培养基上进一步培养,以获得转化和再生的枝条。基因组dna由再生的枝条制备,并且用作模板,以执行用acci的pcr-rflp分析。如图8a中所示,观察到未被acci切割的pcr片段。换言之,获得了在其内iaa9靶序列几乎100%突变的转化和再生的枝条。在14个转化和再生的枝条中,13个枝条显示了与图8a中所示相同的结果。在这些再生的植物中,真叶是单叶状的,其是由iaa9缺乏引起的表型之一。因此,显示了可以通过使用tid的基因组编辑以高效率引入突变。
[表6]
实施例3.高等动物中的基因组编辑
在本实施例中,作为高等动物中的基因组编辑的一个实施方案,证实了本发明的技术在人胚肾细胞衍生的细胞系hek293中有效地起作用。
(1)用于在高等动物细胞中的tid基因表达的载体的构建
基于衍生自来自铜绿微囊藻的tid基因座的cas5d、cas6d、cas7d、cas3d和cas10d的氨基酸序列信息,人工化学合成了编码每种cas蛋白的基因序列。制备了dna片段,其包含核定位信号序列(seqidno:22,seqidno:23),其含有在cas蛋白编码基因各自的5'上游串联排列的两个核定位信号;以及在cas蛋白编码基因之间的自切割肽2a序列(seqidno:24-28)。巨细胞病毒增强子+鸡β-肌动蛋白基因启动子杂合序列(cbh启动子;seqidno:41)在经由2a肽序列彼此融合的五个tid基因片段的5'上游处连接,并且牛生长激素基因终止子序列(bgh终止子;seqidno:42)在经由2a肽序列彼此融合的五个tid基因片段的3'下游处连接,并且从而制备了tid基因表达盒。将tid基因表达盒连接到pcr8topo载体(由thermofisherscientific制造)内,以构建pcr_htid。对于crrna表达盒,人工化学合成了dna(seqidno:31),其中在两个crrna序列之间放置含有两个限制性酶bsai位点的间隔区序列,使得可以连接35个核苷酸的任何序列。作为表达控制序列的人u6snrna基因启动子序列(seqidno:43)在crrna表达盒的5'上游处连接,而聚t序列在crrna表达盒序列的3'下游处连接。将具有人u6snrna基因启动子和聚t序列的crrna表达盒连接到pcr8topo载体(由thermofisherscientific制造)内,以构建pcr_crrna。在pcr_htid中具有核定位信号的编码每种cas蛋白的序列显示为seqidno:33-37。表7中显示了cbh启动子、bgh终止子和人u6snrna基因启动子序列。
[表7]
(2)培养的动物细胞中的基因组编辑
在培养的动物细胞的实施例中,使用人胚肾细胞衍生的细胞系(hek293细胞系),并且选择emx1基因作为用于引入突变的靶序列。在emx1基因中选择靶1(seqidno:44)和靶2(seqidno:45)作为靶序列,并且将靶1和2的人工化学合成的dna连接到用于在上文(1)中制备的培养的人细胞的crrna表达盒内,以构建含有靶1的puc_crrna-t1和含有靶2的puc_crrna-t2。构建的质粒在大肠杆菌菌株hst08(由takarabioinc.制造)中进行扩增,然后使用pureyield(注册商标)plasmidminiprepsystem(由promegacorp.制造)进行纯化。在纯化的质粒中,通过转染将pcr_htid和puc_crrna-t1的混合物或pcr_htid和puc_crrna-t2的混合物引入hek293细胞内。在质粒载体引入后三天,收集细胞,并且使用blood&cellculturednaminikit(由qiagen制造)由其制备基因组dna。使用因此制备的基因组dna作为模板,通过pcr扩增包含靶1或靶2的基因组序列区,并且使用自动电泳仪multina(由shimadzucorporation制造),通过异源双链体迁移率分析执行突变分析。进一步地,将扩增的pcr片段克隆到pneb193载体(由newenglandbiolab制造)内,并且通过测序分析鉴定突变序列。基于“其中观察到突变序列的克隆数目/分析的克隆总数目”来计算体细胞突变效率。质粒未引入其内或者pcr_htid、puc_crrna-t1或puc_crrna-t2已单独引入其内的细胞系用作对照,以相同的方式执行突变分析。关于使用hek293细胞系的基因组编辑的实验方案显示于图9中。
图10和图11显示了当用pcr_htid和puc_crrna-t1的混合物或pcr_htid和puc_crrna-t2的混合物转染hek293细胞系时,或者当未用质粒转染hek293细胞系(对照)时,获得的实验结果。如图10和图11中所示,在用pcr_htid和puc_crrna-t1的混合物或pcr_htid和puc_crrna-t2的混合物转染的hek293细胞系中,检测到指示在靶序列上引入的突变的峰。另一方面,在作为对照的质粒并未引入其内的细胞系中,未检测到指示突变引入的峰。类似于质粒并未引入其内的细胞系,在pcr_htid、puc_crrna-t1或puc_crrna-t2已单独引入其内的细胞系中,未检测到指示突变引入的峰。
然后,将其中通过异源双链体迁移率分析检测到指示突变引入的峰的序列样品克隆到质粒载体内,并且通过测序进行分析。结果,如图12和图13中所示,发现在靶1和靶2上引入了缺失和/或插入突变。
[表8]
工业适用性
根据本发明,能够靶向通过使用crisprii型或v型衍生的rna引导的核酸内切酶的常规基因组编辑技术不能靶向的基因序列。具体地,根据本发明,能够生成突变等位基因,通过转录激活和失活控制基因表达,并且在不能通过常规技术靶向的基因区域上通过dna修饰/组蛋白修饰蛋白质结构域的靶向实现表观基因组改变。
序列表自由文本
seqidno:1;铜绿微囊藻cas5d氨基酸序列
seqidno:2;铜绿微囊藻cas6d氨基酸序列
seqidno:3;铜绿微囊藻cas7d氨基酸序列
seqidno:4;铜绿微囊藻cas3d氨基酸序列
seqidno:5;铜绿微囊藻cas10d氨基酸序列
seqidno:6;含有直接重复(37b)和间隔区(35b的n)的tidcrrna。n是构成关于靶核苷酸序列的互补序列的任何核苷酸
seqidno:7;用于在大肠杆菌中表达的cas5d核苷酸序列
seqidno:8;用于在大肠杆菌中表达的cas6d核苷酸序列
seqidno:9;用于在大肠杆菌中表达的cas7d核苷酸序列
seqidno:10;用于在大肠杆菌中表达的cas3d核苷酸序列
seqidno:11;用于在大肠杆菌中表达的cas10d核苷酸序列
seqidno:12;j23108合成启动子
seqidno:13;核糖体结合序列
seqidno:14;终止子序列stop767
seqidno:15;终止子序列stop768(1)
seqidno:16;终止子序列top768(2)
seqidno:17;t7终止子序列
seqidno:18;crispr重复序列
seqidno:19;t7启动子序列
seqidno:20;crrna表达盒
seqidno:21;合成cccdb基因表达盒
seqidno:22;核定位信号(nls)氨基酸序列
seqidno:23;nls核苷酸序列
seqidno:24;自切割肽2a氨基酸序列
seqidno:25;自切割肽2a(1)编码序列
seqidno:26;自切割肽2a(2)编码序列
seqidno:27;自切割肽2a(3)编码序列
seqidno:28;自切割肽2a(4)编码序列
seqidno:29;2x花椰菜花叶病毒35s基因启动子+ω序列
seqidno:30;拟南芥属休克蛋白18.2kda基因终止子
seqidno:31;crrna表达盒
seqidno:32;拟南芥属u6snrna-26基因启动子序列
seqidno:33;2xnls+cas5d
seqidno:34;2xnls+cas6d
seqidno:35;2xnls+cas7d
seqidno:36;2xnls+cas3d
seqidno:37;2xnls+cas10d
seqidno:38;烟草pds基因上的靶序列1
seqidno:39;烟草pds基因上的靶序列2
seqidno:40;番茄iaa9基因上的靶序列
seqidno:41;巨细胞病毒增强子+通用鸡β-肌动蛋白基因杂合启动子
seqidno:42;牛衍生的生长激素基因终止子序列
seqidno:43;人u6snrna基因启动子
seqidno:44;人emx1基因上的靶1序列
seqidno:45;人emx1基因上的靶2序列。
- 还没有人留言评论。精彩留言会获得点赞!