一种磷酰氧基苯甲醛(或酮)及其衍生物及其制备方法与流程
本发明属于有机合成与多肽化学领域,涉及一种磷酰氧基苯甲醛(或酮)及其衍生物及其制备方法,尤其涉及磷酰氧基苯甲醛(或酮)的还原产物磷酰氧基苯甲醇类以及磷酰氧基苯甲醛(或酮)的肟类衍生物等结构新颖的化合物及其合成制备与分离纯化方法。
背景技术:
氨基和羧基是广泛存在于各类生物活性化合物中的重要官能团,它们的保护与脱保护在现代合成有机化学中通常是必不可少的关键环节,见文献:
[1]i.w.hamley.chem.rev.2017,117,14015-14041.
[2]kocienski,p.j.protectinggroups;georgthiemeverlag:stuttgartnewyork,2004.
[3]green,t.w.;wuts,p.g.m.protectivegroupsinorganicsynthesis;johnwileyandsons:newyork,1999.
尤其是,在多肽化学合成过程中,α-氨基酸的氨基和羧基官能团的保护与脱保护是最重要的问题之一。在肽合成中,一旦氨基酸被活化,就必须防止其聚合或其他副反应的发生。另一方面,肽的合成,无论是在溶液中还是在固相上,都优先从c-端到n-端的方向进行,并且在合成过程中需要反复多次除去α-氨基或羧基上的临时保护基,因此,脱保护的反应必须在不影响侧链残基保护基团或甚至肽链的温和条件下进行,见文献[4]:isidro-llobet,a.;alvarez,m.;albericio,f.chem.rev.2009,109,2455-2504.。
尽管已有数百个保护基团可以通过各种方法产生和除去,但是仍然有必要探索并继续开发新的和温和的策略以引入和切割许多现有的保护基团。
多肽和拟肽药物几乎涉及化学和生物医学的所有学科,见文献:
[5]j.clardyandc.walsh,nature,2004,432,829-837.
[6]h.h.szetoandp.w.schiller,pharm.res.,2011,28,2669–2679.
[7]c.j.whiteanda.k.yudin,nat.chem.,2011,3,509–524.
[8]n.assemanda.k.yudin,nat.protoc.,2012,7,1327–1334.
[9]v.j.hruby,g.li,c.haskellluevanoandm.shenderovich,biopolymers,1997,43,219–266.
这些产品用于学术研究和药物生产的环境友好合成方法的探索已经持续了50多年,见文献:
[10]j.l.gustafson,d.limands.j.miller,science,2010,328,1251–1255.
[11]v.r.pattabiramanandj.w.bode,nature,2011,480,471–479.
[12]e.ko,j.liu,l.m.perez,g.lu,a.schaeferandk.burgess.j.am.chem.soc.,2011,133,462-477.
[13]w.-k.chan,c.-m.ho,m.-k.wongandc.-m.che,j.am.chem.soc.,2006,128,14796-14797.
[14]r.hirschmann,a.b.smithiii,c.m.taylor,etal.science,1994,265,234-237.
[15]a.b.smithiii,a.b.benowitz,p.a.sprengeler,etal.j.am.chem.soc.,1999,121,9286-9298.
80年代已故诺贝尔奖获得者布鲁斯·梅里菲尔德(brucemerrifield)发明的固相肽合成(spps)显示了肽合成的显著成就,见文献:[16]r.b.merrifield.j.am.chem.soc.,1963,85,2149-2154.
这一里程碑式的发现可以克服传统液相肽合成存在纯化步骤繁琐以及消耗大量的原料和试剂(包括偶联剂、溶剂和硅胶)等方面的缺点,可以减少废物的产生;该方法还大大加速了肽和蛋白质的合成与研究。spps还对用于药物发现和开发的一般化学合成施加巨大的影响,特别是对组合化学和高通量筛选,见文献:[17]i.sharmaandd.crich,j.org.chem.,2011,76,6518-6524.
自那时以来,已经开发了几种合成方案以补充spps,以最小化放大的难度,使用过量的试剂和昂贵的树脂用于偶联反应,特别是用于合成较长的多肽和较复杂结构的环肽,见文献:
[18]k.d.eom,z.w.miao,j.l.yangandj.p.tam,j.am.chem.soc.,2003,125,73-82.
[19]s.liu,b.l.penteluteands.b.h.kent,angew.chem.,int.ed.,2012,51,993-999.
[20]d.g.mullen,b.weigel,g.baranyandm.d.distefano,j.pept.sci.,2010,16,219-222.
[21]y.okada,h.suzuki,t.nakae,etal.j.org.chem.,2012,78,320-327.
[22]b.c.li,d.c.montgomery,j.w.puckettandp.b.dervan,j.org.chem.,2013,78,124-133.
[23]k.jin,i.h.sam,k.h.l.po,etal.nat.commn.2016,7,12394
在这方面,利用可溶性聚合物作为氨基酸残基偶联的载体,后来发展了一种替代方法,该方法使得多肽合成能够在液相中进行反应,但是以固相方式,或者通过方便的提取操作来进行分离纯化。然而,后一种方法需要高分子量的可溶性聚合物,这对生产大量分子量比高分子量低得多的小分子肽来说是很不方便,也不符合原子经济的原则。此外,通过仔细控制凝固/结晶条件,通常需要较长的时间来生成固体或结晶产品。
近年来,李桂根教授领导的研究小组在纯化化学领域取得了重大进展,特别关注避免柱层析和重结晶,见文献:
[24]j.wu,g.an,s.lin,j.xie,w.zhou,h.sun,y.pang.li.chem.commun.,2014,50,1259-1261.
[25]c.w.seifert,a.paniagua,g.a.white,l.cai,g.li.eur.j.org.chem.2016,1714-1719.
这一研究和概念被定义为基团辅助纯化(gap)化学/技术。一种有机合成化学,通过有目的地在起始原料或新生产的产品中引入功能良好的基团,避免了传统的纯化方法,如色谱法和/或重结晶法。首先,这项研究有可能涵盖合成有机化学的整个领域。在研究的初始阶段,他们致力于手性胺、n-膦酰基亚胺和n-膦酰亚胺化学的合成,并在这方面取得了很大的成功。通过控制溶解度,可以从粗混合物中选择性地沉淀手性胺产品,从而避免色谱和重结晶。在研究的第二阶段,他们正在开发将此技术扩展到其他底物和官能基团的方法。为此,他们利用了手性助剂的gap性质,并通过修饰,提出了gap保护基的概念。保护基团在几乎每个存在多个官能团的复杂合成中都存在。好的保护基团需要对多种条件具有稳定性,并且必须以高产率添加和除去。gap化学的理想例子是半永久性保护基团导致了gap所必需的溶解特性。问题是,大多数传统的保护基团是非极性的,因此不能产生大多数底物所需的gap溶解度。如果可以开发出能产生足够溶解度控制的保护基团,那么gap化学可以潜在地扩展到所有需要使用该保护基的合成。利用gap技术,无需采用色谱、重结晶等传统纯化方法,即可高效地进行有机合成及三元共沉淀。石油溶剂或共溶剂。因此,可以大大减少原料、硅胶、能源、人力等的使用。gap化学策略可以促进多肽的合成,它具有固相肽合成(spps)和液相肽合成的优点,避免了它们在上述因素方面的缺点。
但是,目前用于氨基酸的氨基或羧基官能团保护的基团,包括上面提及李桂根教授正在研发的gap基团,在脱除过程中都已遭到破坏或分解掉了,完全不能回收再利用,这就大大消耗了生产成本,而且废料的排放必然会造成严重的环境污染问题,无论从经济成本还是从社会效益来看,都还谈不上是最佳方案。所以,在日益提倡经济社会绿色可持续发展理念的今天,探索和发现新的可回收并循环利用的氨基酸羧基保护基团仍然是值得期待重要研究课题。
技术实现要素:
要解决的技术问题
为了避免现有技术的不足之处,本发明提出一种磷酰氧基苯甲醛(或酮)及其衍生物及其制备方法,解决目前多肽化学合成方法中氨基酸保护基团及固相树脂在脱保护或剪切步骤易被破坏或分解,不能直接回收再利用,造成液相合成的反应副产物比较复杂,分离步骤多、耗时周期长、纯化规模小、生产成本高,而固相合成的生产规模小,原料价格贵、浪费大,树脂类废弃物多和环境污染严重的问题。
技术方案
一种磷酰氧基苯甲醛(或酮)及其衍生物,其特征在于:磷酰氧基苯甲醛(或酮)类化合物1的分子结构通式为:
或磷酰氧基苯甲醇类化合物2:
或磷酰氧基苯甲醇类化合物2的fmoc-氨基酸酯类化合物4的分子结构通式:
或磷酰氧基苯甲醛(或酮)肟类化合物3的分子结构通式为:
或磷酰氧基苯甲醛(或酮)肟类化合物3的boc-氨基酸肟酯类化合物5的分子结构通式为:
其中:aa代表各种氨基酸。
一种所述磷酰氧基苯甲醛(或酮)及其衍生物的制备方法,其特征在于步骤如下:
步骤1:采用(ro)2pocl或pocl3与羟基苯甲醛或酮为原料,在a溶剂中加碱催化进行反应,反应完毕后,旋蒸回收溶剂,残留溶液用乙酸乙酯萃取并与水相分离,合并有机相并用去离子水洗涤多次;
然后分离纯化得到磷酰氧基苯甲醛或酮类化合物1;
所述(ro)2pocl或pocl3与羟基苯甲醛或羟基苯甲醛酮的投料比例为1:1~4;
步骤2:以步骤1磷酰氧基苯甲醛或酮类化合物1为原料,以还原剂在b溶剂中进行氢化还原处理反应,反应完毕后,旋蒸回收溶剂,残留溶液用乙酸乙酯萃取并与水相分离,合并有机相并用去离子水洗涤多次;
然后分离纯化得到化合物1相应的衍生化产物磷酰氧基苯甲醇类化合物2;
步骤3:以步骤1得到磷酰氧基苯甲醛或酮类化合物1为原料,与盐酸羟胺在a溶剂中加碱催化进行反应,反应完毕后,旋蒸回收溶剂,残留溶液用乙酸乙酯萃取并与水相分离,合并有机相并用去离子水洗涤多次;
然后分离纯化得到化合物1相应的衍生化产物磷酰氧基苯甲醛或酮肟类化合物3;
步骤4:以步骤2得到的磷酰氧基苯甲醇类化合物2与fmoc保护的氨基酸(fmoc-aa-oh)进行偶联反应,经分离纯化得到化合物2相应的fmoc-氨基酸酯类衍生化产物4;
步骤5:以步骤3得到的磷酰氧基苯甲醛或酮肟类化合物3与boc保护的氨基酸(boc-aa-oh)进行偶联反应,经分离纯化得到化合物3相应的boc-氨基酸肟酯类衍生化产物5。
所述步骤1~步骤5的分离纯化是:采用乙酸乙酯萃取并与水相分离,合并有机相并用去离子水洗涤2~3次;向乙酸乙酯溶液中滴加极性小的烷烃或醚类溶剂,至有结晶或沉淀析出,经过滤和洗涤或重结晶操作完成分离纯化。
所述步骤1~步骤3的加碱催化反应或氢化还原处理反应的反应条件为:在室温下搅拌2~3小时。
所述步骤1~步骤3的用去离子水洗涤多次的次数为2~3次。
所述步骤1和步骤3的a溶剂为:四氢呋喃、乙腈、苯、甲苯、氯仿chcl3、二氯甲烷dcm、n,n-二甲基甲酰胺dmf、n-甲基吡咯烷酮nmp有机溶剂中的一种或几种。
所述步骤1和步骤3的碱为有机碱如叔胺tea,diea,nmm、吡啶及其衍生物dmap中的一种或几种。
所述步骤2的b溶剂为:醇类、乙腈、四氢呋喃等中的一种或几种。
所述步骤2的还原剂为:nabh4、h2/pd/c、b2h4或lialh4。
所述烷烃或醚类溶剂为石油醚、正己烷、环己烷、乙醚、正丁醚等中的一种或几种。
有益效果
本发明提出的一种磷酰氧基苯甲醛(或酮)及其衍生物及其制备方法,(1)采用(ro)2pocl或pocl3与羟基苯甲醛(或酮)为原料,在碱性溶液中进行反应,经分离纯化得到磷酰氧基苯甲醛(或酮)类化合物1;(2)以得到的磷酰氧基苯甲醛(或酮)类化合物1为原料,与盐酸羟胺反应,经分离纯化得到化合物1相应的衍生化产物磷酰氧基苯甲醇类化合物2;(3)以得到的磷酰氧基苯甲醛(或酮)类化合物1为原料,氢化还原处理,经分离纯化得到化合物1相应的衍生化产物磷酰氧基苯甲醛(或酮)肟类化合物3;(4)以得到的磷酰氧基苯甲醇类化合物2与fmoc保护的氨基酸(fmoc-aa-oh)缩合偶联,经分离纯化得到化合物1相应的fmoc-氨基酸酯类衍生化产物4;(5)以得到的磷酰氧基苯甲醛(或酮)肟类化合物3与boc保护的氨基酸(boc-aa-oh)缩合偶联,经分离纯化得到化合物1相应的boc-氨基酸酯类衍生化产物5。
经过系统地筛选研究发现,本发明所涉及的磷酰氧基苯甲醛(或酮)可以与氨基酸的氨基以希夫碱的方式偶联来保护氨基,也很容易经酸解而脱去。另一方面,本发明所涉及的磷酰氧基苯甲醛(或酮)的还原产物(磷酰氧基苯甲醇类衍生物)及其肟化产物(磷酰氧基苯甲醛或酮肟类衍生物)这类小分子在碱性条件和常温常压下经过偶联剂的介导可与氨基酸的羧基发生酯化反应,可高产率地生成稳定的氨基酸酯,用于氨基酸的羧基保护,而且发现磷酸酯类载体及其氨基酸或多肽衍生物在非极性溶剂中很容易结晶而沉淀出来,经简单地分离纯化和碱性水解就可以从其氨基酸或多肽衍生物上将其脱除,副产物还是原来的磷酸酯类载体,回收纯化后可以直接循环利用,实现规模化可持续的绿色生成过程。从现有的文献调研和分析来看,这种利用本发明中所涉及的磷酸酯类载体进行氨基酸羧基保护与辅助纯化的策略尚属首创。与已有的氨基酸羧基保护基团(包括上面提及李桂根教授正在研发的gap基团)或固相树脂载体相比,本发明涉及磷酰氧基苯甲醛(或酮)及其衍生物具有原料丰富易得并可回收再利用、操作简便、条件温和、设备成本低、三废少的绿色环保等优势特色。
本发明所涉及的磷酸酯类小分子在有机合成和多肽化学中具有明显的实际应用价值,既可以作为氨基酸的保护基团,脱保护后不被破坏,容易回收再用。又可以替代固相多肽合成中的树脂载体,利于辅助多肽的分离纯化,剪切后不被破坏,容易回收再用。兼备了液相和固相合成法的优点,可以更加简便、快捷、节约、高效地合成制备保护的氨基酸或多肽及其衍生物,而且磷酸酯类载体可以回收并直接再利用,降低原材料浪费,减少废弃物污染,节约成本,利于环保。
附图说明
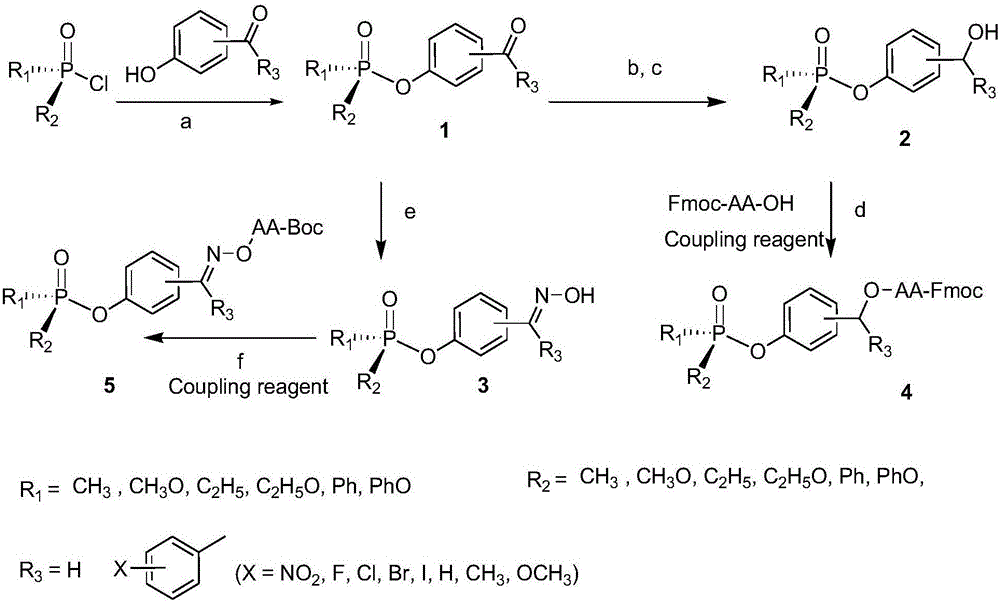
图1:一种磷酰氧基苯甲醛(或酮)及其衍生物的制备方法实例流程图
具体实施方式
现结合实施例、附图对本发明作进一步描述:
实施例主要包括如下化合物及其制备步骤:(1)采用(ro)2pocl或pocl3与羟基苯甲醛(或酮)为原料,在碱性溶液中进行反应,经分离纯化得到磷酰氧基苯甲醛(或酮)类化合物1;(2)以得到的磷酰氧基苯甲醛(或酮)类化合物1为原料,与盐酸羟胺反应,经分离纯化得到化合物1相应的衍生化产物磷酰氧基苯甲醇类化合物2;(3)以得到的磷酰氧基苯甲醛(或酮)类化合物1为原料,氢化还原处理,经分离纯化得到化合物1相应的衍生化产物磷酰氧基苯甲醛(或酮)肟类化合物3;(4)以得到的磷酰氧基苯甲醇类化合物2与fmoc保护的氨基酸(fmoc-aa-oh)缩合偶联,经分离纯化得到化合物1相应的fmoc-氨基酸酯类衍生化产物4;(5)以得到的磷酰氧基苯甲醛(或酮)肟类化合物3与boc保护的氨基酸(boc-aa-oh)缩合偶联,经分离纯化得到化合物1相应的boc-氨基酸酯类衍生化产物5。本发明的详细合成路线见图1。本发明中一些常用的缩写具有以下含义:
boc:叔丁氧羰基
dcm:二氯甲烷ch2cl2
dmap:4-二甲氨基吡啶
dmf:n,n-二甲基甲酰胺
edc-hcl:1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐
fmoc:芴甲氧羰基
gps:绿色多肽合成载体
gsh:谷胱甘肽
hatu:2-(7-氧化苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸盐
hobt:1-羟基苯并三唑
hbtu:o-苯并三氮唑-四甲基脲六氟磷酸盐
nmm:n-甲基吗啉
nmp:n-甲基吡咯烷酮
pybop:六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷
tbu:叔丁基
tfa:三氟乙酸
thf:四氢呋喃
trt:三苯甲基
本发明适用于磷酰氧基苯甲醛(或酮)及其衍生物的制备,反应原理与技术路线见图1。
实际操作步骤
4-二苯基磷酰氧基二苯甲酮(dbk,1a)的合成:准确称取4-羟基二苯甲酮(5g,25mmol,1equiv)于250ml的反应瓶中,加入100ml的四氢呋喃thf并置于冰浴中搅拌30min,反应体系中滴加缚酸剂三乙胺et3n(4.2ml,30mmol,1.2equiv),量取取二苯基次磷酰氯(5.7ml,30mmol,1.2equiv)逐滴加入上述反应体系,置于冰浴中反应30min后,移去冰浴tlc检测反应体系室温反应1.5h,加入稀硫酸(0.1mol/l,10ml)淬灭反应,旋转蒸发仪浓缩除去thf溶剂并加入20ml去离子水,加入乙酸乙酯萃取得到有机相,无水硫酸镁干燥。旋蒸至干后,加入2ml的乙酸乙酯是样品充分溶解,逐滴加入正己烷14ml(vea/vn-hexane=1:7),体系中出现大量白色沉淀,过滤白色沉淀并干燥得到目标化合物(1a),产率约93%。结构表征:1hnmr(400mhz,cdcl3),δ=7.95-7.90(m,4h),7.76-7.73(m,4h),7.59-7.55(m,3h),7.52-7.44(m,6h),7.36-7.34(d,2h)ppm;31pnmr(162mhz,cdcl3),δ=31.51ppm;13cnmr(100mhz,cdcl3),δ=195.4,154.4,137.5,133.9,132.8,132.4,132.1,131.8,131.7,131.2,129.9,128.8,128.7,128.3,120.5ppm;hrms(esi)m/zcalcdforc25h20o3p+(m+h)+=399.11446,found399.11469。
所述反应体系中的溶剂可以用四氢呋喃thf、乙腈、苯、甲苯、氯仿chcl3、二氯甲烷dcm、n,n-二甲基甲酰胺dmf、n-甲基吡咯烷酮nmp有机溶剂中的一种或几种。
碱为有机碱如叔胺tea,diea,nmm、吡啶及其衍生物dmap中的一种或几种。
烷烃或醚类溶剂为石油醚、正己烷、环己烷、乙醚、正丁醚等中的一种或几种。
4-二苯基磷酰氧基二苯甲醇(dbm,2a)的合成:准确称取4-二苯基磷酰氧基二苯甲酮(1a)(800mg,2mmol,1equiv)于100ml的反应瓶中,加入甲醇溶液20ml置于冰浴中搅拌30min,反应体系分三批次加入硼氢化钠nabh4(92mg,2.4mmol,1.2equiv),加上气球转入室温密封反应2h,tlc检测反应至原料消耗完毕后加入饱和氯化铵淬灭反应,浓缩除去甲醇溶液后加入去离子水10ml,乙酸乙酯萃取得到有机相,无水硫酸镁干燥。旋蒸至干后,加入0.5ml的乙酸乙酯是样品充分溶解,逐滴加入正己烷5ml(vea/vn-hexane=1:10),体系中出现大量白色沉淀,过滤白色沉淀并干燥得到目标化合物(2a),产率约97%。结构表征:1hnmr(400mhz,cdcl3),δ=7.89-7.84(m,4h),7.56-7.52(m,2h),7.48-7.43(m,4h),7.31-7.23(m,7h),7.14-7.12(d,2h),5.74(s,1h),2.99(s,1h)ppm;31pnmr(162mhz,cdcl3),δ=30.58ppm;13cnmr(100mhz,cdcl3),δ=150.0,143.9,140.6,132.5,131.8,131.7,130.1,128.7,128.6,128.4,128.0,127.4,126.6,120.6,75.4ppm;hrms(esi)m/zcalcdforc25h22o3p+(m+h)+=401.13011,found:401.12985。
所述反应体系中的溶剂可以用醇类、乙腈、四氢呋喃等中的一种或几种。
还原剂为:nabh4、h2/pd/c、b2h4或lialh4。
烷烃或醚类溶剂为石油醚、正己烷、环己烷、乙醚、正丁醚等中的一种或几种。
4-二苯基磷酰氧基二苯甲酮肟(dbo,3a)的合成:准确称取4-二苯基磷酰氧基二苯甲酮(dbk,1a)(800mg,2mmol,1equiv)于100ml的反应瓶中,加入50ml的无水乙醇溶液,搅拌加入5ml的吡啶(etoh:pyridine=10:1),随后加入nh2oh·hcl(280mg,4mmol,2equiv),室温搅拌10h,反应结束后浓缩除去乙醇溶剂及部分的吡啶,随后加入50ml乙酸乙酯溶解,加入稀hcl连续洗涤两次,除净残留吡啶及过量的羟胺,最后使用无水硫酸镁干燥2h,旋干后加入0.5ml的乙酸乙酯使样品充分溶解,逐滴加入正己烷4ml(vea/vn-hexane=1:8),体系中出现大量白色沉淀,过滤白色沉淀并干燥得到目标化合物(3a),产率约92%。结构表征:1hnmr(400mhz,cdcl3),δ=9.50(s,1h),7.99-7.90(m,4h),7.59-7.28(m,14h),7.22-7.20(m,1h)ppm;31pnmr(162mhz,cdcl3),δ=31.09ppm;13cnmr(100mhz,cdcl3),δ=156.5,151.3,136.4,132.7,131.9,131.3,130.0,129.8,129.3,129.0,128.8,128.6,128.3,128.0,120.4ppm;hrms(esi)m/zcalcdforc25h22o3p+(m+h)+401.13011,found:401.29789。
所述反应体系中的溶剂可以用四氢呋喃thf、乙腈、苯、甲苯、氯仿chcl3、二氯甲烷dcm、n,n-二甲基甲酰胺dmf、n-甲基吡咯烷酮nmp有机溶剂中的一种或几种。
碱为有机碱如叔胺tea,diea,nmm、吡啶及其衍生物dmap中的一种或几种。
烷烃或醚类溶剂为石油醚、正己烷、环己烷、乙醚、正丁醚等中的一种或几种。
4-二苯基磷酰氧基二苯甲基酪氨酸酯[dbm-o-tyr(otbu)-fmoc,4a]的合成:准确称取4-二苯基磷酰氧基二苯甲醇(dbm,2a)(200mg,0.5mmol,1equiv)于100ml的反应瓶中,加入30mldcm溶解,反应体系依次加入fmoc-tyr(otbu)-oh(276mg,0.6mmol,1.2equiv),4-二甲氨基吡啶dmap(7.32mg,0.06mmol,0.12equiv),二环己基碳二亚胺dcc(123mg,0.6mol,1.2equiv),室温下反应2h,tlc检测反应结束后冷却至0℃后过滤得到滤液,浓缩后加入乙酸乙酯30ml溶解,依次用饱和nh4cl水溶液、饱和na2co3溶液洗涤,无水硫酸镁干燥。旋蒸浓缩后用1ml的乙酸乙酯溶解样品,逐滴加入正己烷6ml(vea/vn-hexane=1:6),体系中出现大量白色沉淀,过滤白色沉淀并干燥得到目标化合物dbm-o-tyr(tbu)-fmoc(4a),产率约95%,经过一次沉淀可作为原料进行下一步投料,连续洗涤2~3次样品可进行nmr表征。结构表征:1hnmr(400mhz,cdcl3),δ=7.93-7.88(m,4h),7.80-7.78(d,2h),7.59-7.54(m,4h),7.50-7.40(m,6h),7.33-7.20(m,11h),6.87(s,1h),6.82-6.79(d,4h),5.32-5.29(d,1h),4.79-4.74(m,1h),4.44-4.32(m,2h),4.23-4.19(m,1h),3.16-3.10(m,2h),1.34(s,9h)ppm;31pnmr(162mhz,cdcl3),δ=30.86ppm;13cnmr(100mhz,cdcl3),δ=170.6,155.6,154.5,150.5,143.8,141.3,139.1,135.7,132.6,131.8,130.2,129.8,129.1,128.7,128.6,128.1,127.7,127.1,125.1,124.1,120.8,120.0,78.4,67.0,54.8,47.2,37.4,33.9,28.9ppm;hrms(esi)m/zcalcdforc53h49no7p+(m+h)+=842.32412,found:842.32422.
4-二苯基磷酰氧基二苯甲酮肟boc-缬氨酸酯(dbo-o-val-boc,5a)的合成:准确称取4-二苯基磷酰氧基二苯甲酮肟(dbo,3a)(206mg,0.5mmol,1equiv)于100ml的反应瓶中,加入30mldcm溶解,反应体系依次加入boc-val-oh(130mg,0.6mmol,1.2equiv),4-二甲氨基吡啶dmap(7.32mg,0.06mmol,0.12equiv),二环己基碳二亚胺dcc(123mg,0.6mol,1.2equiv),室温下反应2h,tlc检测反应结束后冷却至0℃后过滤得到滤液,浓缩后加入乙酸乙酯30ml溶解,依次用饱和nh4cl水溶液、饱和nahco3溶液洗涤,无水硫酸镁干燥,浓缩后用0.5ml的乙酸乙酯溶解样品,逐滴加入正己烷6ml(vea/vn-hexane=1:12),体系中出现大量白色沉淀,过滤白色沉淀并干燥得到化合物gap-c=n-o-val-boc(5a),产率约98%,经过一次沉淀可作为原料进行下一步投料,连续洗涤2~3次样品可进行nmr表征。结构表征:1hnmr(400mhz,cdcl3),δ=7.97-7.87(m,4h),7.61-7.21(m,15h),5.04-5.02(m,1h),4.20-4.17(m,1h),1.95-1.90(m,1h),1.45(s,9h),0.88-0.77(m,6h)ppm;31pnmr(162mhz,cdcl3),δ=30.66ppm;13cnmr(100mhz,cdcl3),δ=169.9,164.9,155.5,151.9,134.2,132.7,132.1,131.8,131.7,131.2,130.6,129.8,129.1,128.8,128.7,128.4,128.3,120.5,79.9,57.7,31.3,28.3,18.8,17.6ppm;hrms(esi)m/zcalcdforc35h37n2o6p+(m+h)+613.23892,found:613.23795。
4-二苯基磷酰氧基二苯甲酮肟缬氨酸酯(dbo-o-val-fmoc,5a’)的合成:准确称取4-二苯基磷酰氧基二苯甲酮肟(dbo,3a)(206mg,0.5mmol,1equiv)于100ml的反应瓶中,加入30mldcm溶解,反应体系依次加入fmoc-val-oh(203mg,0.6mmol,1.2equiv),4-二甲氨基吡啶dmap(7.32mg,0.06mmol,0.12equiv),二环己基碳二亚胺dcc(123mg,0.6mol,1.2equiv),室温下反应2h,tlc检测反应结束后冷却至0℃后过滤得到滤液,浓缩后加入乙酸乙酯30ml溶解,依次用饱和nh4cl水溶液、饱和nahco3溶液洗涤,无水硫酸镁干燥,浓缩后用0.5ml的乙酸乙酯溶解样品,逐滴加入正己烷6ml(vea/vn-hexane=1:12),体系中出现大量白色沉淀,过滤白色沉淀并干燥得到化合物gap-c=n-o-val-fmoc(5a’),产率约95%,经过一次沉淀可作为原料进行下一步投料,连续洗涤2~3次样品可进行nmr表征。结构表征:1hnmr(400mhz,cdcl3),δ=7.95-7.89(m,4h),7.79-7.78(d,2h),7.62-7.25(m,21h),5.36-5.31(m,1h),4.43-4.39(m,2h),4.29-4.23(m,2h),2.01-1.99(m,1h),0.98-0.80(m,6h)ppm;31pnmr(162mhz,cdcl3),δ=30.56ppm;13cnmr(100mhz,cdcl3),δ=169.6,165.2,156.1,152.1,143.7,141.3,134.1,132.7,131.8,131.7,131.3,130.7,130.6,129.8,129.2,128.8,128.7,128.5,128.3,127.7,127.1,125.1,120.7,120.0,67.1,58.2,47.2,31.6,22.7,17.6ppm;hrms(esi)m/zcalcdforc45h39n2o6p+(m+h)+735.25457,found:735.25534。
- 还没有人留言评论。精彩留言会获得点赞!