用于富集CRISPR/Cas9介导的同源重组修复细胞的筛选报告载体及筛选方法与流程
本发明属于基因工程技术领域,涉及可用于富集crispr/cas9介导的同源重组修复细胞的筛选报告载体的构建和应用。
背景技术:
crispr/cas9核酸酶技术是继zfns和talens之后的第三代靶向编辑技术。jinek等人在2012年6月首次报道利用来源于酿脓链球菌(s.pyogenes)ii型crispr/cas9系统(clusteredregularlyinterspacedshortpalindromicrepeats(crispr)/crispr-associatedprotein-9nuclease(cas9))的蛋白和导向rna(guiderna,grna)可以体外靶向切割质粒dna。crispr/cas9系统利用一个cas9核酸酶和包含20nt靶序列的导向rna(sgrna)可以对基因组特定位点产生dna双链断裂(doublestrandbreaks,dsbs)。dsbs能激活细胞内的多种dna断裂修复机制,主要有不依赖于同源序列的非同源末端连接(non-homologousendjoining,nhej),以及同源序列介导的同源重组(homologousrecombination,hr)。nhej修复机制是哺乳动物细胞中dsbs修复的主要机制,即在细胞内既没有供体dna以及在dsb两边又没有正向重复序列的情况下,断裂dna分子两端会通过非特异性的末端连接实现修复,同时一般会引入少量核苷酸碱基的插入或缺失(insertionsanddeletions,indels),进而可能导致移码突变并引起基因破坏甚至是基因敲除。同源重组修复(homology-directedrepair,hdr)是在有供体dna存在的情况下,断裂dna分子可以通过同源重组机制来修复,从而产生精确的定点替换、插入突变体、精确删除启动子或外显子等dna序列。然而,hdr修复效率远低于nhej修复,这限制了其在精准基因组工程中的广泛应用。由此可见,提高hdr效率是基因组编辑研究中不懈努力的目标和方向。
目前为止,研究报道开发了许多行之有效的提高hdr效率的策略,主要方式包括抑制nhej通路(如抑制ligaseiv、ku70/80蛋白活性)、强化hdr通路(如增加rad51/rad52的表达或者使用激动剂brefeldina和l755507),以及改变细胞周期(如延长s和g2期)。另外,dna供体模板的结构,包括长的同源臂供体、线性双链供体、单链寡核苷酸(ssodn)供体、双切供体(doublecutdonors)都可以提高hdr效率。
除了较低的hdr效率,从所有处理的细胞中鉴定出正确编辑的细胞也是复杂繁琐并且耗费时间的过程。最初的富集方式是通过质粒带有的筛选标记基因(例如荧光蛋白或者抗生素基因)来筛选转染阳性的细胞,这种富集方法只是筛选转染质粒表达的细胞。随后开发的筛选报告载体基于nhej或者ssa修复机制,是通过筛选进行核酸切割的细胞来富集。两种机制的筛选报告载体更适合富集有插入缺失(indels)的细胞。目前常用的富集hdr编辑细胞的方式是通过在靶位点处插入筛选标记基因表达盒来筛选,但是这种方法需要进一步将编辑细胞中的筛选标记去除。近两年研究报道了基于“共打靶筛选”的(co-targetingwithselection)策略,即在基因组上一个不相关的位点上通过hdr修复方式插入筛选标记基因来筛选,可以提高另外一个或者多个位点基于hdr编辑的效率,从而可以高效富集易于hdr编辑的细胞。然而,该方法还是会导致编辑细胞的基因组上不相干位点处筛选表达盒的固有整合,并对细胞功能产生影响。
技术实现要素:
本发明的目的在于提供一种用于富集crispr/cas9介导的同源重组修复细胞的筛选报告载体及筛选方法。
为达到上述目的,本发明采用了以下技术方案:
一种crispr/cas9介导的筛选报告载体(hdr-usr),包括通用(universal)sgrna表达盒、cas9表达盒、抗性基因表达盒以及抗性基因同源重组修复模板,所述通用(universal)sgrna表达盒的转录区域包括在基因编辑目标细胞的基因组上无可识别的靶序列的导向rna(即该导向rna识别的靶位点与所述细胞的基因组不同源),抗性基因表达盒的转录区域包括与抗性基因编码区同源的上游片段、与抗性基因编码区同源的下游片段以及位于该上、下游片段之间的由所述导向rna识别的靶序列。
优选的,所述基因编辑目标细胞选自哺乳动物细胞。
优选的,所述导向rna识别的靶序列选自如seq.id.no.1所示的序列或该序列的同源序列。
优选的,所述上游片段与所述抗性基因编码区中自起始位点向下游延伸的一段序列对应,所述下游片段与所述抗性基因编码区中自终止位点向上游延伸的一段序列对应;所述上、下游片段的片段大小之和小于等于所述抗性基因编码区的大小。hdr-usr主要借助由所述导向rna识别的靶序列及靶序列下游pam区引入的移码突变阻止所述抗性基因表达,进一步可以结合抗性基因编码区的片段缺失,从而保证抗性基因表达盒仅在自身经过同源重组修复后表达抗性基因,当hdr-usr共转染至目标细胞后,若其抗性基因表达通过同源重组修复恢复,则在与hdr-usr共转染的crispr/cas9基因编辑供体载体及打靶载体作用下同时发生基因组上目标位置同源重组修复的细胞,能够通过抗性筛选得到富集。
优选的,所述抗性基因同源重组修复模板选自不包含起始密码子的抗性基因编码区对应序列,从而保证其不表达抗性基因。
优选的,所述抗性基因选自puro,puro经同源重组修复后表达嘌呤霉素(puromycin)抗性,通过用嘌呤霉素对共转染后的细胞进行药物筛选,可以加快对基因组发生同源重组修复的细胞的富集。
上述筛选报告载体的构建方法,包括以下步骤:
1)以抗性基因序列(例如,包含在nhej-rpg质粒中的puro编码区序列)为模板,采用重叠pcr扩增得到上述抗性基因表达盒的转录区域;将该转录区域连接至第一骨架载体(例如,pcdna3.1),得到抗性基因表达盒克隆质粒;
2)将抗性基因表达盒以及扩增(例如,以包含在nhej-rpg质粒中的puro编码区序列为模板)得到的抗性基因同源重组修复模板(例如,δpuro)连接至第二骨架载体(例如,pxl-bacii),得到含有修复模板的抗性基因表达盒克隆质粒;
3)将上述导向rna识别的靶序列(如t1,参见seq.id.no.1)连接至含有cas9表达盒的第三骨架载体(例如,px330-u6-chimeric_dbbsi-cbh-hspcas9),得到通用(universal)sgrna表达盒及cas9表达盒的克隆质粒;
4)将通用(universal)sgrna表达盒及cas9表达盒连接至含有修复模板的抗性基因表达盒克隆质粒,得到筛选报告载体(hdr-usr)。
一种crispr/cas9介导的同源重组修复细胞的筛选方法,包括以下步骤:
1)构建针对细胞基因组hdr编辑位点的crispr/cas9打靶载体及相对应的供体载体;
2)将构建得到的crispr/cas9打靶载体及相对应的供体载体与上述筛选报告载体(hdr-usr)共转染细胞;
3)转染48~72小时后,在转染体系中添加用于筛选能够表达所述抗性基因(例如,puro)的细胞的药物(例如,嘌呤霉素);
4)经所述药物筛选3~5天后,将筛选得到的细胞(此即为富集的细胞)转入正常细胞培养基(不含药物)中进行细胞克隆培养。
优选的,所述药物的添加终浓度为0.2~5μg/ml(根据不同细胞系对抗生素的实际抗性而异)。
优选的,所述培养具体包括以下步骤:将上述筛选得到的细胞稀释后培养细胞单克隆(例如,铺到96孔板培养皿,培养7天);然后通过pcr分型鉴定得到经过hdr编辑的阳性细胞单克隆;将阳性细胞单克隆进行扩大培养(例如,转至24孔板)。
上述筛选报告载体在富集crispr/cas9介导的同源重组修复细胞中的应用。
本发明的有益效果体现在:
本发明提出的筛选报告载体(hdr-usr)自身可以通过抗性基因(例如,puro)序列同源重组修复而使所转染细胞产生抗性,通过共转染该载体及对转染体系进行对应药物(例如,嘌呤霉素)筛选,即可实现对基因组发生hdr编辑的细胞的富集(其本质是提高了所处理细胞池的hdr效率),显著提高了hdr编辑细胞的筛选效率。且由于所述筛选报告载体(hdr-usr)仅是自身发生hdr,在细胞基因组hdr编辑中也无需对细胞基因组的其他位置再引入hdr,因此,在达到富集hdr编辑细胞的同时,不会对细胞基因组的不相关位点产生双链断裂与dna整合,为推动精准基因编辑的研究和应用提供有效途径。
将本发明所述的筛选报告载体(hdr-usr)与用于细胞基因组hdr编辑的打靶载体及供体载体组成共转染系统,可应用于高效富集发生点编辑、片段插入和片段删除的hdr阳性细胞克隆,并且可广泛应用于哺乳动物的多种细胞。
附图说明
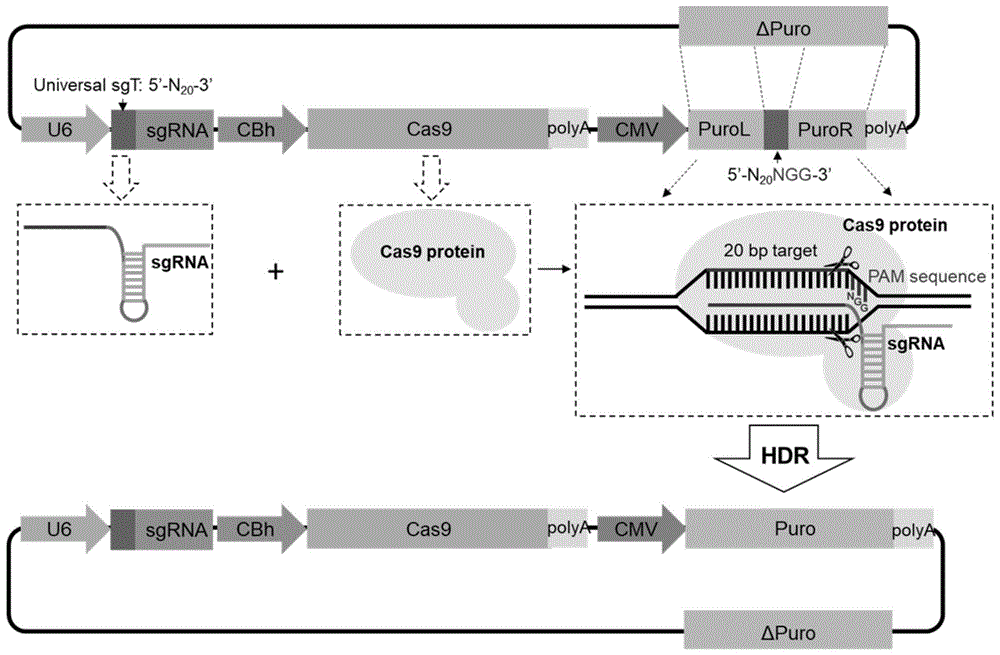
图1为hdr-usr载体工作示意图。
图2为purol-t1_ngg-puror片段琼脂糖凝胶电泳图。
图3为pcdna3.1-cmv-purol-t1_ngg-puror-polya载体酶切鉴定琼脂糖凝胶电泳图。
图4为cmv-purol-t1_ngg-puror-polya片段琼脂糖凝胶电泳图。
图5为pxl-cmv-purol-t1_ngg-puror-polya载体酶切鉴定琼脂糖凝胶电泳图。
图6为δpuro片段琼脂糖凝胶电泳图。
图7为pxl-cmv-purol-t1_ngg-puror-polya-δpuro载体酶切鉴定琼脂糖凝胶电泳图。
图8为px330-sgt1-cbh-hspcas9载体酶切鉴定琼脂糖凝胶电泳图。
图9为u6-sgt1-cbh-cas9-polya片段琼脂糖凝胶电泳图。
图10为hdr-usr载体酶切鉴定琼脂糖凝胶电泳图。
图11为hdr-usr载体图谱。
图12为利用基于hdr-usr的共转染系统富集hdr编辑细胞的操作流程。
图13为pd-emx1的酶切鉴定琼脂糖凝胶电泳图。
图14为对照组与试验组ecori酶切检测图。
图15为通过facs分析egfpknock-in效率的结果。
图16为通过pcr扩增(a)和xbai酶切(b)检测试验组hdr效率的结果。
具体实施方式
以下结合附图和实施例对本发明作进一步的详细说明。所述实施例仅用于解释本发明,而非对本发明保护范围的限制。
(一)hdr-usr载体构建
本发明提出了一种用于富集crispr/cas9介导的hdr编辑细胞的筛选报告载体hdr-usr(crispr/cas9mediatedhdr-universalsurrogatereporter)。
hdr-usr主要由四个部分组成,分别是:universalsgrna表达盒、cas9表达盒、被universalsgrna对应的靶序列打断的puro表达盒,以及去除atg的puro编码序列(即δpuro)。hdr-usr转染进入细胞后,可以表达universalsgrna和cas9蛋白;之后cas9蛋白在universalsgrna的指导下对puro表达盒中puro左臂(purol)和puro右臂(puror)中间的靶序列区切割,同时可以利用载体上带有的δpuro序列对打断的puro左臂(purol)和puro右臂(puror)进行hdr修复,形成完整的puro编码区,从而在cmv的启动下实现puro抗性的表达,参见图1。
(一)筛选报告载体hdr-usr的构建
1.靶位点的选择
预设计的hdr-usr载体内部的靶位点不会打靶哺乳动物细胞基因组,于是选择文献报道的sirnanc序列,将u修改为t(参见以下靶序列的下划线部分)后,在序列5’端加碱基g作为靶序列,另外pam区选择ngg(例如,ggg)。即hdr-usr中表达的universalsgrna(sgt1)所含的靶序列(t1)为(2018年3月设计完成):
5’-gttctccgaacgtgtcacgt-3’
根据http://crispr.mit.edu/网站预测该序列的靶位点活性得分96分,并在人和小鼠基因组无打靶,脱靶效应很低。
2.构建pcdna3.1-cmv-purol-t1_ngg-puror-polya载体
(1)引物设计与合成:设计overlappcr扩增purol-t1_ngg-puror的引物。引物序列如下(2018年3月设计完成):
puro-f(nhei):5’-ctagctagcatgaccgagtacaagcccacg-3’
puro-r(+t1):5’-ccttctccgcttgtggcattcgctccagaccgccaccgcgg-3’
puro-f(+t1):5’-gcgaatgccacaagcggagaaggaggcctcctggcgccgcac-3’
puro-r(apai):5’-aatgggccctcaggcaccgggcttgcgg-3’
其中,小写字母为内切酶识别位点,设计的引物送南京金斯瑞生物科技有限公司进行合成,下同。
(2)overlappcr扩增purol-t1_ngg-puror片段:以nehj-rpg(renetal.,cellmollifesci,2015,72(14):2763-2772.)为模板,用puro-f(nhei)与puro-r(+t1),及puro-f(+t1)与puro-r(apai)分别扩增位于puro编码区(cdna,共597bp)的片段一(含puro编码区第1至第229bp的cdna序列,即purol)及片段二(含puro编码区第330至第597bp的cdna序列,即puror);以片段一和片段二为模板,用puro-f(nhei)与puro-r(apai)扩增purol-t1_ngg-puror片段(541bp)。pcr产物经1%琼脂糖凝胶电泳检测片段大小正确,结果如图2所示。胶回收所得片段。
(3)片段与骨架载体酶切:purol-t1_ngg-puror片段与pcdna3.1载体(优宝生物,产品编号vt1001)分别用nhei和apai双酶切,胶回收酶切后的片段和骨架。酶切体系如表1:
表1.酶切反应体系
(4)连接:将酶切后的片段和pcdna3.1载体骨架用t4连接酶连接,连接体系如表2。16℃连接过夜,然后转化大肠杆菌dh5α感受态细胞(天根生化,产品编号cb101),涂lb/amp平板,挑取单克隆并在lb/amp液体培养基中37℃培养8h。
表2.连接反应体系
(5)酶切检测鉴定阳性克隆:不同单克隆的菌液提质粒,用nhei和apai双酶切鉴定。阳性克隆酶切后条带为5322bp+533bp(参见图3),即pcdna3.1-cmv-purol-t1_ngg-puror-polya载体。
3.构建pxl-cmv-purol-t1_ngg-puror-polya载体
(1)引物设计与合成:设计扩增cmv-purol-t1_ngg-puror-polya片段的引物。引物序列如下(2018年3月设计完成):
cmv-f(noti):5’-aatgcggccgccgatgtacgggccagatatac-3’
polya-r(bglii):5’-ggaagatctgagccccagctggttctttc-3’
(2)cmv-purol-t1_ngg-puror-polya片段的扩增:以第2步构建的pcdna3.1-cmv-purol-t1_ngg-puror-polya载体为模板,用引物cmv-f(noti)与polya-r(bglii)扩增cmv-purol-t1_ngg-puror-polya片段(1522bp),电泳结果如图4所示。
(3)片段与骨架载体酶切:cmv-purol-t1_ngg-puror-polya片段与pxl-bacii载体(wuetal.,febsletters,2017,591(6):903-913)分别用noti与bglii双酶切,胶回收酶切后的片段和pxl-bacii载体骨架。酶切体系参考表1。
(4)连接:将酶切后的片段和骨架用t4连接酶连接,连接体系参考表2。16℃连接过夜,然后转化大肠杆菌dh5α感受态细胞,涂lb/amp平板,挑取单克隆并在lb/amp液体培养基中37℃培养8h。
(5)酶切检测鉴定阳性克隆:不同单克隆的菌液提质粒,用noti与bglii双酶切鉴定。阳性克隆酶切后条带为4046bp+1506bp(参见图5),即pxl-cmv-purol-t1_ngg-puror-polya载体。
4.构建pxl-cmv-purol-t1_ngg-puror-polya-δpuro载体
(1)引物设计与合成:设计扩增δpuro片段的引物。引物序列如下(2018年3月设计完成):
δpuro-f(kpni):5’-cggggtaccaccgagtacaagcccacggtgc-3’
δpuro-r(hindiii):5’-cccaagcttggcaccgggcttgcgggt-3’
(2)扩增δpuro片段:以nhej-rpg为模板,用δpuro-f(kpni)与δpuro-r(hindiii)扩增不含有atg(起始密码子)的puro基因编码区δpuro(puro编码区第4至第597bp的cdna序列)。结果如图6所示(扩增片段大小为612bp)。
(3)片段与骨架载体酶切:δpuro片段和pxl-cmv-purol-t1_ngg-puror-polya载体分别用kpni和hindiii双酶切(位于cmv-purol-t1_ngg-puror-polya下游),胶回收酶切后的片段和载体骨架。酶切体系参考表1。
(4)连接转化与提质粒、酶切鉴定操作参考步骤3,kpni和hindiii酶切鉴定结果参见图7,阳性克隆酶切后条带为5520bp+596bp,即pxl-cmv-purol-t1_ngg-puror-polya-δpuro载体。
5.构建px330-sgt1-cbh-hspcas9载体
(1)引物设计与合成:设计靶序列的拟合引物,插入px330-u6-chimeric_dbbsi-cbh-hspcas9载体(addgeneplasmid#42230)的bbsi之间。
拟合引物序列如下(2018年3月设计完成):
univsal-t1-f1(bbsi):5’-caccgttctccgaacgtgtcacgt-3’
univsal-t1-r1(bbsi):5’-aaacacgtgacacgttcggagaac-3’
(2)引物退火拟合:引物univsal-t1-f1(bbsi)和univsal-t1-r1(bbsi)稀释为10pmol,各吸取5μl,混匀,置于pcr仪95℃变性10min,自然冷却至室温退火拟合,获得带有bbsi粘性末端的bs双链。
(3)骨架载体酶切:px330-u6-chimeric_dbbsi-cbh-hspcas9载体用bbsi酶切3~4h(酶切体系参见表3),胶回收骨架8484bp。
表3.酶切反应体系
(4)连接:将bs双链与回收载体骨架用t4连接酶连接(连接体系参见表4),16℃连接过夜,然后转化大肠杆菌dh5α感受态细胞,涂lb/amp平板,挑取单克隆并在lb/amp液体培养基中37℃培养8h。
表4.连接反应体系
(5)鉴定阳性克隆:不同单克隆的菌液提质粒,用bbsi和saci双酶切鉴定。阳性克隆酶切后条带为6580bp+1929bp(参见图8),即px330-sgt1-cbh-hspcas9载体。
6.构建pxl-u6-sgt1-cbh-cas9-polya-cmv-purol-t1_ngg-puror-polya-δpuro(即hdr-usr)
(1)引物设计与合成:设计引物扩增u6-sgt1-cbh-cas9-polya片段,并插入pxl-cmv-purol-t1_ngg-puror-polya-δpuro载体的bamhi和noti之间(位于cmv-purol-t1_ngg-puror-polya上游)。
引物序列如下(2018年3月设计完成):
u6-f(bamhi):5’-cgcggatcctttcccatgattccttcatat-3’
bgh-r(noti):5’-aatgcggccgctccccagcatgcctgc-3’
(2)u6-sgt1-cbh-cas9-polya片段的扩增:以构建好的px330-sgt1-cbh-hspcas9为模板,用u6-f(bamhi)与bgh-r(noti)扩增u6-sgt1-cbh-cas9-polya片段(片段长度约5800bp,图9)。胶回收所得片段。
(3)片段与骨架载体酶切:u6-sgt1-cbh-cas9-polya片段与pxl-cmv-purol-t1_ngg-puror-polya-δpuro载体分别用bamhi和noti双酶切。
(4)连接转化与提质粒酶切鉴定操作参考步骤3,bamhi和hindiii酶切鉴定结果参见图10。阳性克隆酶切后条带为7902bp+3958bp,即hdr-usr载体。
7.报告载体hdr-usr的图谱
构建好的hdr-usr如图11所示(11860bp)。hdr-usr包括顺次排列的universalsgrna表达盒(具体包括u6启动子、t1及grnascaffold)、cas9表达盒(具体包括cbh启动子、cas9编码区及polya)、抗性基因puro表达盒(具体包括cmv启动子、抗性基因puro编码区同源序列purol、t1、pam区序列ggg、抗性基因puro编码区同源序列puror及polya)以及抗性基因同源重组修复模板(δpuro),其中,polya简称pa。
(二)hdr-usr作为报告载体的工作原理及流程
工作原理:hdr-usr自身可以通过同源重组修复而产生嘌呤霉素抗性,通过共转染该载体并通过嘌呤霉素药物筛选富集hdr编辑的细胞。
具体的富集试验过程:
1)设计细胞基因组中目标编辑位点的crispr/cas9打靶载体及相对应的供体载体;
2)将构建好的crispr/cas9打靶载体、供体载体和hdr-usr共转染细胞;
3)转染48小时后,在细胞培养基中添加嘌呤霉素(puromycin);
4)用嘌呤霉素筛选5天后,撤药,换为正常细胞培养基,得到经过筛选后的细胞,即实现富集细胞;
5)将经过筛选后的细胞用有限稀释法稀释后铺到96孔板培养皿;
6)7天后,随机筛选细胞单克隆,用pcr鉴定基因组目标编辑位点发生hdr编辑的阳性细胞单克隆(hdr编辑细胞);
7)将阳性单克隆细胞转至24孔板,对细胞进行扩大培养。
参见图12,以上2)-7)步中,采用含有10%胎牛血清及100μg/ml青/链霉素的dmem培养基为细胞培养基,并于37℃、co2浓度为5%的细胞培养箱中进行培养。
(三)hdr-usr作为报告载体在不同位点富集crispr/cas9介导的hdr点编辑、片段插入和片段删除中的具体应用
3.1以人emx1位点为例,通过hdr将emx1靶序列的pam区突变为ecori酶切位点,利用hdr-usr系统来富集精确点编辑(pointmutation)的细胞(hdr编辑细胞)。
1.构建emx1位点的crispr/cas9打靶载体px330-emx1-sgrna
(1)首先选择emx1靶位点(gagtccgagcagaagaagaaggg),设计靶位点的拟合引物。引物序列如下(2018年4月设计完成):
emx1-f(bbsi):5’-caccgagtccgagcagaagaagaa-3’
emx1-r(bbsi):5’-aaacttcttcttctgctcggactc-3’
(2)引物emx1-f(bbsi)和emx1-r(bbsi)稀释为10pmol,各吸取5μl,混匀,置于pcr仪95℃变性10min,自然冷却至室温退火拟合,获得带有bbsi粘性末端的bs双链。
(3)px330-u6-chimeric_dbbsi-cbh-hspcas9骨架载体用bbsi酶切,胶回收。
(4)拟合产物与回收骨架连接,转化,提质粒送公司进行测序,构建得到px330-emx1-sgrna。
2.构建点突变供体pxl-emx1donor(pd-emx1)
(1)以hek293t细胞(购自中国科学院细胞库)基因组为模板,用emx1donor-f1(bamhi)与emx1donor-r1,及emx1donor-f2与emx1donor-r2(sacii)分别扩增片段一及片段二,以片段一和片段二为模板,用emx1donor-f1(bamhi)和emx1donor-r2(sacii)扩增bamhi-emx1donor-sacii片段。引物序列如下(2018年4月设计完成,下划线部分为引入突变的位置):
emx1donor-f1(bamhi):5’-cgcggatccttctcctgactgttccttgtg-3’
emx1donor-r1:5’-ggttgatgtgatgggaattcttcttcttctgctcggactc-3’
emx1donor-f2:5’-gaattcccatcacatcaaccggtggcgcattgccacgaag-3’
emx1donor-r2(sacii):5’-tccccgcgggcacgttgctctttcttgggct-3’
(2)bamhi-emx1donor-sacii片段与pxl-bacii载体分别用bamhi和sacii双酶切,胶回收目的片段。
(3)连接,转化,提质粒酶切鉴定(图13),阳性克隆酶切后条带为5106bp+1045bp,即pd-emx1载体。
3.构建瞬时筛选对照组质粒prs426-cmv-t2a-egfp-polya(简称ppuro-t2a-egfp)
(1)以nhej-rpg为模板,利用puro-f(bamhi)与puro-r(nhei)引物扩增puro编码区片段,引物如下(2018年4月设计完成):
puro-f(bamhi):5’-cgcggatcccatgaccgagtacaagcccac-3’
puro-r(nhei):5’-ctagctagcggcaccgaaaaaaggcttgcgggt-3’
(2)puro编码区片段与prs426-cmv-egfp载体(yanetal.,fishandshellfishimmunology,2015,47(2):758-765)分别用bamhi和nhei双酶切,胶回收目的片段。
(3)连接,转化,提质粒酶切鉴定,送阳性质粒到公司测序,构建得到ppuro-t2a-egfp。
4.将px330-emx1-sgrna、pd-emx1与hdr-usr载体共转染hek293t细胞作为试验组(hdr-usr组)。同时,将px330-emx1-sgrna与pd-emx1共转染hek293t细胞作为空对照组(blank组),将px330-emx1-sgrna、pd-emx1和ppuro-t2a-egfp共转染hek293t细胞作为puro瞬筛对照组(ppuro-t2a-egfp组)。每个组设置三个重复。
5.试验组转染48小时后,向puro瞬筛对照组与试验组中添加嘌呤霉素(3μg/ml)对细胞进行药物筛选。
6.blank组细胞转染72小时后收集细胞混合池部分细胞;puro瞬筛对照组与试验组用药物筛选5天后,撤药,更换为正常细胞培养基,收集细胞混合池部分细胞。提取三组细胞的基因组dna。
7.以三个组的dna为模板,用检测引物emx1-detection-f1与emx1-detection-r1(2018年4月设计完成)来扩增靶序列区(2138bp),胶回收后用ecori酶切检测。未编辑的基因组不能被eocri酶切开,精确编辑后的基因组可以被ecori酶切开,条带大小为1087bp+1060bp。
emx1-detection-f1:5’-cactgccagacacagaatagg-3’
emx1-detection-r1:5’-gcacgttgctctttcttgggct-3’
8.利用imagej软件对酶切后的电泳条带进行灰度分析,根据灰度分析结果检测三个组的同源重组效率。结果如图14所示,可以看出,共转染hdr-usr的试验组相对于blank组hdr效率提高了9倍以上,共转染hdr-usr的试验组相对于puro瞬筛对照组hdr效率提高了5倍以上,即hdr-usr可以用于高效富集基因组发生hdr的细胞。
3.2以人gapdh位点为例,通过hdr在gapdh终止密码子之前插入t2a-egfp-polya序列,利用hdr-usr富集crispr/cas9介导的片段插入(knock-in)的hdr编辑细胞。
1.构建gapdh-3’utr区的crispr/cas9打靶载体px330-gapdh-sgrna
(1)设计gapdh靶序列拟合引物,序列如下(2018年5月设计完成):
hugapdh-sg1-f1(bbsi):5’-caccggggagattcagtgtggtgg-3’
hugapdh-sg1-r1(bbsi):5’-aaacccaccacactgaatctcccc-3’
(2)构建过程参考上例(3.1)
2.构建egfpdonor质粒pxl-gapdhlha-t2a-egfp-polya-gapdhrha(简称pd-egfp-ki)
(1)首先将质粒prs426-cmv-egfp用bamhi和kpni双酶切,将t2a-gfp-polya片段切下,插入pxl-bacii的bamhi和kpni酶切位点之间,构建得到pxl-t2a-gfp-polya。
(2)以hek293t细胞基因组为模板,用gapdh-lha-f(bamhi)与gapdh-lha-r(nhei)扩增gapdh-lha,胶回收,用bamhi和nhei双酶切,插入pxl-t2a-gfp-polya的bamhi和nhei双酶切位点之间,构建得到pxl-gapdh-lha-t2a-gfp-polya。引物序列如下(2018年5月设计完成):
gapdh-lha-f(bamhi):5’-cgcggatcctctcttagatttggtcgtattggg-3’
gapdh-lha-r(nhei):5’-ctagctagcctccttggaggccatgtgg-3’
(3)以hek293t细胞基因组为模板,用gapdh-rha-f(kpni)与gapdh-rha-r(clai)扩增gapdh-rha,用kpni和clai双酶切,插入pxl-gapdh-lha-t2a-gfp-polya的kpni和clai酶切位点之间,连接,构建pxl-gapdhlha-t2a-egfp-polya-gapdhrha,即pd-egfp-ki。引物序列如下(2018年5月设计完成):
gapdh-rha-f(kpni):5’-cggggtacctcctcacagttgccatgtagacc-3’
gapdh-rha-r(clai):5’-cccatcgatggacctttcctccatccctgt-3’
3.将px330-gapdh-sgrna和pd-egfp-ki共转染hek293t细胞,作为对照组;
px330-gapdh-sgrna、pd-egfp-ki和hdr-usr载体共转染hek293t细胞,作为试验组。每个组设置六孔板中的三个孔为重复。
4.试验组转染48小时后,向试验组中添加嘌呤霉素(3μg/ml)对细胞进行药物筛选。
5.对照组转染72小时后收集细胞,试验组药物筛选5天后收集细胞。收集的细胞池进行流式荧光活性细胞分选(fluorescence-activatedcellsorting,facs)统计,统计出发绿色荧光的细胞数(egfp+)和总细胞数,egfp+细胞数与细胞总数的比值作为片段插入的效率。分析结果参见图15,可以看出,共转染hdr-usr的试验组相对于对照组hdr效率提高了4.5倍,即富集了hdr编辑细胞。
3.3以人aavs1位点为例,通过hdr精确删除aavs1基因组序列(约1kb),删除后的序列连接处形成xbai酶切位点,利用hdr-usr富集crispr/cas9介导的片段删除的的hdr编辑细胞。
1.构建aavs1两个靶位点的crispr/cas9打靶载体px330-aavs1-sg1和px330-aavs1-sg2
(1)设计两个位点的拟合引物,序列如下(2018年6月设计完成):
aavs1-5-target-f(bbsi):5’-caccgccagcgagtgaagacggcat-3’
aavs1-5-target-r(bbsi):5’-aaacatgccgtcttcactcgctggc-3’
aavs1-3-target-f(bbsi):5’-caccgcaggtaaaactgacgcacgg-3’
aavs1-3-target-r(bbsi):5’-aaacccgtgcgtcagttttacctgc-3’
(2)载体构建过程参考上例(3.1)
2.构建pxl-aavs1deletedonor(pd-aavs1deletion)
(1)以hek293t细胞基因组为模板,用引物aavs1(deletedonor)-f1(ecori)与aavs1(delete)-r1,及aavs1(delete)-f2与aavs1(deletedonor)-r2(hindiii)分别扩增片段一及片段二。然后以片段一和片段二为模板,用引物aavs1(deletedonor)-f1(ecori)和aavs1(deletedonor)-r2(hindiii)扩增aavs1deletedonor片段。引物序列如下(2018年5月设计完成):
aavs1(deletedonor)-f1(ecori):5’-ccggaattctggactccaccaacgccgac-3’
aavs1(delete)-r1:5’-tggctactggccttatctagagatgcccggagaggacccag-3’
aavs1(delete)-f2:5’-tctagataaggccagtagccagccccgtcct-3’
aavs1(deletedonor)-r2(hindiii):5’-cccaagcttccttccgccctcagatgaag-3’
(2)aavs1deletedonor片段用ecori和hindiii双酶切,插入pxl-bacii的ecori和hindiii酶切位点之间,构建pd-aavs1deletion。
3.将px330-aavs1-sg1、px330-aavs1-sg2和pd-aavs1deletion共转染hek293t细胞,作为对照组;px330-aavs1-sg1、px330-aavs1-sg2,pd-aavs1deletion和hdr-usr载体共转染hek293t细胞,作为试验组。每个组设置六孔板中的三个孔为重复。
4.试验组转染48小时后,向试验组中添加嘌呤霉素(3μg/ml)对细胞进行药物筛选。
5.对照组细胞转染72小时后收集细胞混合池部分细胞;试验组用药物筛选5天后,撤药,收集细胞混合池部分细胞。提取两组细胞的基因组dna。
6.以两个组的dna为模板,用检测引物aavs1-detection-f1与aavs1-detection-r1来扩增靶序列区(2138bp),胶回收后用xbai酶切检测。引物序列如下:
aavs1-detection-f1:5’-agccggtcctggactttgtct-3’
aavs1-detection-r1:5’-cccaacccctccccattcaac-3’
如果未发生片段删除,细胞基因组扩出长度为2899bp的片段,如果发生片段删除(约1kb),细胞基因组扩出长度约1880bp的片段。对于扩出的删除型条带(1880bp),如果精确编辑后的基因组可以被xbai酶切开,条带大小为1357bp+523bp。检测结果如图16,可以看出,经过筛选后,pcr扩增对照组基因组,只扩增出未删除型条带(2899bp),扩增试验组只扩增出删除型条带(1880bp),因此,利用hdr-usr富集细胞后,获得基因组大片段删除的效率显著提高,经进一步xbai酶切鉴定,利用hdr-usr富集后的细胞,hdr介导的片段删除效率可以达到41.8%。
<110>西北农林科技大学
<120>用于富集crispr/cas9介导的同源重组修复细胞的筛选报告载体及筛选方法
<160>40
<210>1
<211>20
<212>dna
<213>t1
<400>1
gttctccgaacgtgtcacgt20
<210>2
<211>30
<212>dna
<213>puro-f(nhei)
<400>2
ctagctagcatgaccgagtacaagcccacg30
<210>3
<211>41
<212>dna
<213>puro-r(+t1)
<400>3
ccttctccgcttgtggcattcgctccagaccgccaccgcgg41
<210>4
<211>42
<212>dna
<213>puro-f(+t1)
<400>4
gcgaatgccacaagcggagaaggaggcctcctggcgccgcac42
<210>5
<211>28
<212>dna
<213>puro-r(apai)
<400>5
aatgggccctcaggcaccgggcttgcgg28
<210>6
<211>32
<212>dna
<213>cmv-f(noti)
<400>6
aatgcggccgccgatgtacgggccagatatac32
<210>7
<211>29
<212>dna
<213>polya-r(bglii)
<400>7
ggaagatctgagccccagctggttctttc29
<210>8
<211>31
<212>dna
<213>δpuro-f(kpni)
<400>8
cggggtaccaccgagtacaagcccacggtgc31
<210>9
<211>27
<212>dna
<213>δpuro-r(hindiii)
<400>9
cccaagcttggcaccgggcttgcgggt27
<210>10
<211>24
<212>dna
<213>univsal-t1-f1(bbsi)
<400>10
caccgttctccgaacgtgtcacgt24
<210>11
<211>24
<212>dna
<213>univsal-t1-r1(bbsi)
<400>11
aaacacgtgacacgttcggagaac24
<210>12
<211>30
<212>dna
<213>u6-f(bamhi)
<400>12
cgcggatcctttcccatgattccttcatat30
<210>13
<211>27
<212>dna
<213>bgh-r(noti)
<400>13
aatgcggccgctccccagcatgcctgc27
<210>14
<211>23
<212>dna
<213>emx1靶位点
<400>14
gagtccgagcagaagaagaaggg23
<210>15
<211>24
<212>dna
<213>emx1-f(bbsi)
<400>15
caccgagtccgagcagaagaagaa24
<210>16
<211>24
<212>dna
<213>emx1-r(bbsi)
<400>16
aaacttcttcttctgctcggactc24
<210>17
<211>30
<212>dna
<213>emx1donor-f1(bamhi)
<400>17
cgcggatccttctcctgactgttccttgtg30
<210>18
<211>40
<212>dna
<213>emx1donor-r1
<400>18
ggttgatgtgatgggaattcttcttcttctgctcggactc40
<210>19
<211>40
<212>dna
<213>emx1donor-f2
<400>19
gaattcccatcacatcaaccggtggcgcattgccacgaag40
<210>20
<211>31
<212>dna
<213>emx1donor-r2(sacii)
<400>20
tccccgcgggcacgttgctctttcttgggct31
<210>21
<211>30
<212>dna
<213>puro-f(bamhi)
<400>21
cgcggatcccatgaccgagtacaagcccac30
<210>22
<211>33
<212>dna
<213>puro-r(nhei)
<400>22
ctagctagcggcaccgaaaaaaggcttgcgggt33
<210>23
<211>21
<212>dna
<213>emx1-detection-f1
<400>23
cactgccagacacagaatagg21
<210>24
<211>22
<212>dna
<213>emx1-detection-r1
<400>24
gcacgttgctctttcttgggct22
<210>25
<211>24
<212>dna
<213>hugapdh-sg1-f1(bbsi)
<400>25
caccggggagattcagtgtggtgg24
<210>26
<211>24
<212>dna
<213>hugapdh-sg1-r1(bbsi)
<400>26
aaacccaccacactgaatctcccc24
<210>27
<211>33
<212>dna
<213>gapdh-lha-f(bamhi)
<400>27
cgcggatcctctcttagatttggtcgtattggg33
<210>28
<211>28
<212>dna
<213>gapdh-lha-r(nhei)
<400>28
ctagctagcctccttggaggccatgtgg28
<210>29
<211>32
<212>dna
<213>gapdh-rha-f(kpni)
<400>29
cggggtacctcctcacagttgccatgtagacc32
<210>30
<211>30
<212>dna
<213>gapdh-rha-r(clai)
<400>30
cccatcgatggacctttcctccatccctgt30
<210>31
<211>25
<212>dna
<213>aavs1-5-target-f(bbsi)
<400>31
caccgccagcgagtgaagacggcat25
<210>32
<211>25
<212>dna
<213>aavs1-5-target-r(bbsi)
<400>32
aaacatgccgtcttcactcgctggc25
<210>33
<211>25
<212>dna
<213>aavs1-3-target-f(bbsi)
<400>33
caccgcaggtaaaactgacgcacgg25
<210>34
<211>25
<212>dna
<213>aavs1-3-target-r(bbsi)
<400>34
aaacccgtgcgtcagttttacctgc25
<210>35
<211>29
<212>dna
<213>aavs1(deletedonor)-f1(ecori)
<400>35
ccggaattctggactccaccaacgccgac29
<210>36
<211>41
<212>dna
<213>aavs1(delete)-r1
<400>36
tggctactggccttatctagagatgcccggagaggacccag41
<210>37
<211>31
<212>dna
<213>aavs1(delete)-f2
<400>37
tctagataaggccagtagccagccccgtcct31
<210>38
<211>29
<212>dna
<213>aavs1(deletedonor)-r2(hindiii)
<400>38
cccaagcttccttccgccctcagatgaag29
<210>39
<211>21
<212>dna
<213>aavs1-detection-f1
<400>39
agccggtcctggactttgtct21
<210>40
<211>21
<212>dna
<213>aavs1-detection-r1
<400>40
cccaacccctccccattcaac21
- 还没有人留言评论。精彩留言会获得点赞!