PS-b-PMA(Az)嵌段共聚物及其制备方法和应用与流程
本发明涉及新型高分子材料技术领域,尤其涉及ps-b-pma(az)嵌段共聚物及其制备方法和应用。
背景技术:
嵌段共聚物(blockcopolymer,bcp)是由两种或两种以上性质不同的聚合物链段通过化学键连接一起的特殊聚合物。嵌段结构的不同使得bcp相分离形成形貌丰富的周期性纳米结构。其中,ab两嵌段共聚物是最简单的一类。嵌段共聚物嵌段间的不相容性(用flory-huggins参数表示,χab),使得共聚物体系发生相分离。单从bcp结构来说,聚合度(n,即重复单元数)决定了相分离的有效尺寸。而bcp中的两相a或者b的体积含量(f)决定了最终的自组装形貌。根据两相成分所占比例(f)的不同,大致有球形、柱形、层状以及规整的双连续、双金刚石结构。基于上述理论,一种“自下而上”的嵌段共聚物薄膜自组装微纳加工技术逐渐发展起来,并成为目前光刻技术的研究热点。
一般在嵌段共聚物薄膜自组装微纳加工技术中,两嵌段聚合物旋涂在支撑体上形成超薄的聚合物薄膜。根据bcp与支撑体间的表面相互作用,形成平行或者垂直取向的纳米结构。其中,聚苯乙烯-b-聚甲基丙烯酸甲酯(ps-b-pmma)具有较大的光刻差异性,被广泛应用在光刻领域。通过180~200度几个小时的高温热退火处理,ps-b-pmma能在中立的聚合物刷上形成有序垂直取向纳米结构,极大满足了微电子领域产业化需求。然而,ps-b-pmma之间的χab很小,在300k时χab仅为0.06。这导致该体系难以得到微相分离尺寸小于20nm的图案模板。同时,ps-b-pmma的两相与支撑体间的界面能较大,很难在较宽的薄膜厚度范围内得到垂直柱状或者层状结构。只能通过对支撑体表面进行修饰(如,在旋涂bcp薄膜前在支撑体上制备一层合适无规聚合物刷、化学预图案法和外场调控法等)来制备高度有序的垂直层状和柱状结构图案。
2002年,iyoda课题组用原子转移自由基聚合(atrp)法制备了一系列的peo基液晶侧链型嵌段共聚物。通过侧链液晶基团的引入,bcp能形成膜厚为几个纳米到几个微米高度有序的小尺寸柱状相分离薄膜结构。同时,peo具有一定的吸电子能力,常被用来与含有不同侧链液晶结构单元的聚甲基丙烯酸酯类形成嵌段共聚物,液晶侧链种类如偶氮苯类/二苯基乙烯类/亚苄基苯胺/苯基苯乙烯酮类等。这种peo基侧链液晶bcp模板也被广泛用来制备无机或者金属纳米材料。但是,peo与偶氮液晶的刻蚀差异性不大,该类bcp在纳米光刻领域难以得到进一步推广。
因此,为了满足光刻领域需求,借助ps和pmma优异光刻差异性,在pmma嵌段部分引入大χ值的偶氮液晶侧链来提高bcp的χab,从而实现20nm以下更小尺寸的纳米光刻图案。该嵌段聚合物(ps-b-pma(az))在bcp光刻领域的发展和应用上具有重要的意义。
技术实现要素:
本发明的目的在于提供ps-b-pma(az)嵌段共聚物及其制备方法和应用。为了实现上述目的,本发明的技术方案之一是:一种ps-b-pma(az)嵌段共聚物,具有如下结构:
其中,x为0-15;m为10-800;n为2-200。
本发明的x取值优选为1-11,m取值优选为50-200或25-100或10-100,n取值优选为2-100或5-70或45-70。
在优选的技术方案中,所述嵌段共聚物中x=0,m=100,n=20;或x=6,m=100,n=23;或x=11,m=28,n=68;或x=11,m=42,n=16;或x=11,m=60,n=15;或x=11,m=100,n=44;或x=11,m=100,n=22;或x=11,m=100,n=18。
在另一种优选的技术方案中,所述嵌段共聚物中x=11,m=100,n=44。
优选地,本发明所述的ps-b-pma(az)嵌段共聚物,其ps的体积含量fps在0.001-0.9之间,优选在0.001~0.7之间。
本发明的技术方案之二是:一种ps-b-pma(az)嵌段共聚物的制备方法,具体包括如下步骤:
(1)以小分子引发剂、配体、苯乙烯、氯化亚铜为原料,经原子转移自由基聚合反应得到ps大分子引发剂;
(2)以所述ps大分子引发剂、所述配体、偶氮苯单体、氯化亚铜为原料,经原子转移自由基聚合反应得到ps-b-pma(az)嵌段共聚物。
优选地,所述小分子引发剂选自α-溴代异丁酸乙酯,1-氯苯基乙烷,α-溴代苯乙烷,α-溴代异丁酸甲酯,α-氯代异丁酸乙酯,α-氯代异丁酸甲酯中的一种;优选α-溴代异丁酸乙酯。
优选地,步骤(1)和(2)中,采用的配体相同,所述配体选自三(2-二甲氨基乙基)胺(me6tren)、五甲基二乙烯三胺(pmdeta)、五甲基二丙烯三胺中的一种,优选为pmdeta。
优选地,步骤(2)中,所述偶氮苯单体(含有不同烷基柔性间隔基)选自{11-[4-(4-丁基苯偶氮)苯氧基]十五烷基甲基丙烯酸酯}或ma(az)-0~14c中的一种;其中x的限定同上文对所述ps-b-pma(az)嵌段共聚物的限定(如0~15,优选1-11等)。
优选地,步骤(1)在溶剂中进行,所述溶剂选自氯苯、n,n-二甲基甲酰胺(dmf)中的一种或二者的混合物;为了保证反应的顺利进行,本发明优选除水溶剂作为反应溶剂,即优选无水氯苯,无水dmf中的一种或二者的混合物,更优选无水氯苯;当采用二者的混合物作为反应溶剂时,二者的用量比无特殊限制。
本发明优先选择无水氯苯作为合成ps大分子引发剂的溶剂,该溶剂在室温下能很好溶解小分子引发剂、配体、苯乙烯单体等,使得atrp聚合为均相体系,反应进行得更彻底。同时,在该溶剂下引发剂能保持高的活性,其引发单体聚合得到的ps均聚物多分散性(pdi)小,利于下一步atrp更好进行。
优选地,步骤(2)在溶剂中进行,所述溶剂选自四氢呋喃、氯苯、n,n-二甲基甲酰胺(dmf)中的一种或二者或三者的混合物。同样为了保证反应的顺利进行,本发明优选无水溶剂作为反应溶剂,即优选无水四氢呋喃、无水氯苯、无水n,n-二甲基甲酰胺中的一种或二者或三者的混合物,更优选无水氯苯;当选用2种或2种以上的混合溶剂时,各溶剂的用量无特殊限制。
本发明优先选择无水氯苯作为合成ps-b-pma(az)的溶剂,该溶剂在室温下能很好溶解聚苯乙烯大分子引发剂、ma(az)-0~15c单体、ps-b-pma(az)聚合物等,使得atrp聚合为均相体系,反应进行得更彻底。同时,在该溶剂下引发剂能保持高的活性,其引发单体聚合得到的bcp多分散性(pdi)小,bcp自组装结构缺陷更少。
优选地,以摩尔比计,步骤(1)中,小分子引发剂:配体:氯化亚铜(cucl):苯乙烯(st)=1:(1-3):(1-2):(10-900)。
优选地,以摩尔比计,步骤(2)中,大分子引发剂:配体:氯化亚铜(cucl):偶氮苯单体=1:(1-3):(1-2):(0.1-110)。
优选地,步骤(1)和(2)的反应时间均为10-20h,进一步优选为16h。
作为本发明较佳的ps-b-pma(az)嵌段共聚物的制备方法,步骤(1)中所用的原料为α-溴代异丁酸乙酯、pmdeta、st和cucl,步骤(2)中所用的原料为ps大分子引发剂、pmdeta、ma(az)和cucl,其余atpr操作同上,此种情况下,能够实现ps基液晶嵌段共聚物的atrp的高效合成,可得到不同嵌段数窄分布的大分子引发剂和两亲性液晶嵌段共聚物,实现了ps的可控atrp合成。
更优选地,本发明所述制备方法包括如下步骤:
(1)ps大分子引发剂的合成:将小分子引发剂、配体、苯乙烯(st)、溶剂加入反应容器,用液氮冷冻所述反应器,并加入氯化亚铜(cucl);在液氮冷冻状态下抽真空3-7min,再在通氮气条件下解冻并搅拌3-7min,重复上述抽真空、解冻操作2-5次,最后,于真空状态下,90-120℃反应6-72h,即得粗产品;
(2)ps-b-pma(az)嵌段共聚物的合成:将ps大分子引发剂、配体、偶氮苯单体(ma(az)-0~15c)、溶剂加入反应容器,采用液氮冷冻所述反应器,并加入氯化亚铜;在液氮冷冻状态下抽真空3-7min,再在通氮气情况下解冻并搅拌3-7min,重复上述抽真空、解冻操作2-5次,最后,于真空状态下,90-120℃反应6-72h,即得粗产品;
其中,所述“解冻”指的是将反应容器拿出液氮环境。
为了确保产物的品质,上述制备方法还包括对步骤(1)和(2)的反应液进行后处理的步骤,对两步反应的后处理可采用相同的操作,即:用液氮淬灭反应,去除铜盐,旋干反应液并用醚类和醇类溶剂沉淀、洗涤产物;
其中,所述醚类溶剂为石油醚、乙醚或二者的混合物;优选采用冷的醚类溶剂对产物进行沉淀、洗涤。
优选地,采用中性氧化铝柱去除铜盐。进一步优选地,在去除铜盐之前,采用二氯甲烷溶解反应产物。
本发明同时提供上述任意一种方法制备得到的ps-b-pma(az)嵌段共聚物。
本发明合成的ps-b-pma(az)嵌段共聚物是由两步atrp反应聚合而成。聚合过程中严格排气处理,包括:合理规范的操作、多次冷冻解冻操作、加入的所有溶剂均通氮气排气处理,从而成功地实现了ps基液晶嵌段共聚物的atrp有效合成。
本发明的技术方案之三是:一种ps-b-pma(az)嵌段共聚物光刻图案薄膜,所述薄膜由上述任一种ps-b-pma(az)嵌段共聚物或上述任一种方法制备而成的ps-b-pma(az)嵌段共聚物制得。
优选地,所述ps-b-pma(az)嵌段共聚物光刻图案薄膜呈垂直柱状或垂直层状结构。
优选地,所述垂直柱状薄膜柱直径为几纳米到几十纳米。
优选地,所述垂直层状薄膜层间距为几纳米到几十纳米。
本发明所述ps-b-pma(az)嵌段共聚物光刻图案薄膜呈垂直柱状或垂直层状结构,所述垂直柱状薄膜柱直径或所述垂直层状薄膜层间距可以调节;例如选择ps的体积含量fps=0.001-0.9之间不同m值的嵌段共聚物即可得到柱直径为几个纳米到几十个纳米的垂直柱状薄膜,或层间距为几个纳米到几十个纳米的垂直层状薄膜。
本发明的技术方案之四是:一种ps-b-pma(az)嵌段共聚物光刻图案薄膜的制备方法,具体包括如下步骤:将配制好的ps-b-pma(az)嵌段共聚物溶液旋涂在支撑体上形成一定厚度的薄膜,对所述薄膜进行热退火和反应离子刻蚀(rie)即得。
所述ps-b-pma(az)嵌段共聚物为上述任意一种ps-b-pma(az)嵌段共聚物,或由上述任意一种方法制备得到。
为了确保涂膜的效果,在进行涂膜之前,先对支撑体进行清洗,清洗可采用本领域常规技术手段,本发明优选的清洗操作为:将支撑体先后分别置于丙酮和乙醇中超声清洗,然后用氮气吹扫干燥。
优选地,所述支撑体为硅片、聚对苯二甲酸乙二醇酯(pet)、铝箔中的一种。
优选地,以氯仿和/或甲苯为溶剂配制浓度为0.5wt%-5wt%的所述ps-b-pma(az)嵌段共聚物溶液。
优选地,所述旋涂具体为:将配置好的溶液滴在支撑体上,在1000rpm-5000rpm的涂膜转速下旋涂成膜,控制膜厚为100nm-500nm,涂膜后置于室温下干燥。
优选地,所述退火的条件为:于100-200℃下退火2-30min;更优选于120-150℃下退火5-10min。
优选地,利用o2、ar混合气对热退火后的聚合物薄膜进行刻蚀,所述混合气的体积流量为5~40/5~10sccm,刻蚀功率为30~100w,刻蚀时间为10~50s;更优选地,所述体积流量为40/10sccm,刻蚀功率为30~50w,刻蚀时间为10~30s。
本发明制备得到的ps-b-pma(az)嵌段共聚物光刻图案薄膜退火后的薄膜厚度能达到100nm~500nm,退火后能清楚看到薄膜截面呈高度有序的ps六方柱状分散相结构和pma(az)为连续相的垂直柱状结构;以及垂直层状结构。
本发明的技术方案之五是:上述任意一种ps-b-pma(az)嵌段共聚物光刻图案薄膜或上述任意一种方法制备得到的ps-b-pma(az)嵌段共聚物光刻图案薄膜在微电子领域中的应用;优选在无机纳米材料制备中的应用。
本发明所述的嵌段共聚物中,一个嵌段为聚{11-[4-(4-丁基苯偶氮)苯氧基]烷基甲基丙烯酸酯}(pma(az)-0~15c),具有易相分离和光降解(偶氮基团)性质;另一嵌段为聚苯乙烯(ps),具有较快的离子刻蚀速率的特点;嵌段共聚物中两嵌段间相互作用哈金斯参数值较大,且含有稳定相分离的偶氮嵌段,容易实现模板特征尺寸在20纳米以下的需求,为嵌段共聚物光刻、金属纳米线的制备、集成电路板的制备提供了潜在的共聚物材料。bcp薄膜模板能够形成高度有序的垂直柱状和层状图案形貌。刻蚀掉其中的pma(az)相后,再结合原子沉积技术,可用来制备得到结构完整长程有序的半导体、过渡金属、金属纳米线等材料。
本发明涉及的“ps-b-pma(az)”、“ps-b-pma(az)两嵌段共聚物”、“ps-b-pma(az)液晶嵌段共聚物”、“ps-b-pma(az)嵌段共聚物”、“ps-b-pma(az)共聚物”指代的含义相同。
本发明涉及到的原料或试剂均可市购获得。
在符合本领域常识的基础上,上述各优选条件可以相互组合,即得本发明各较佳实施例。
附图说明
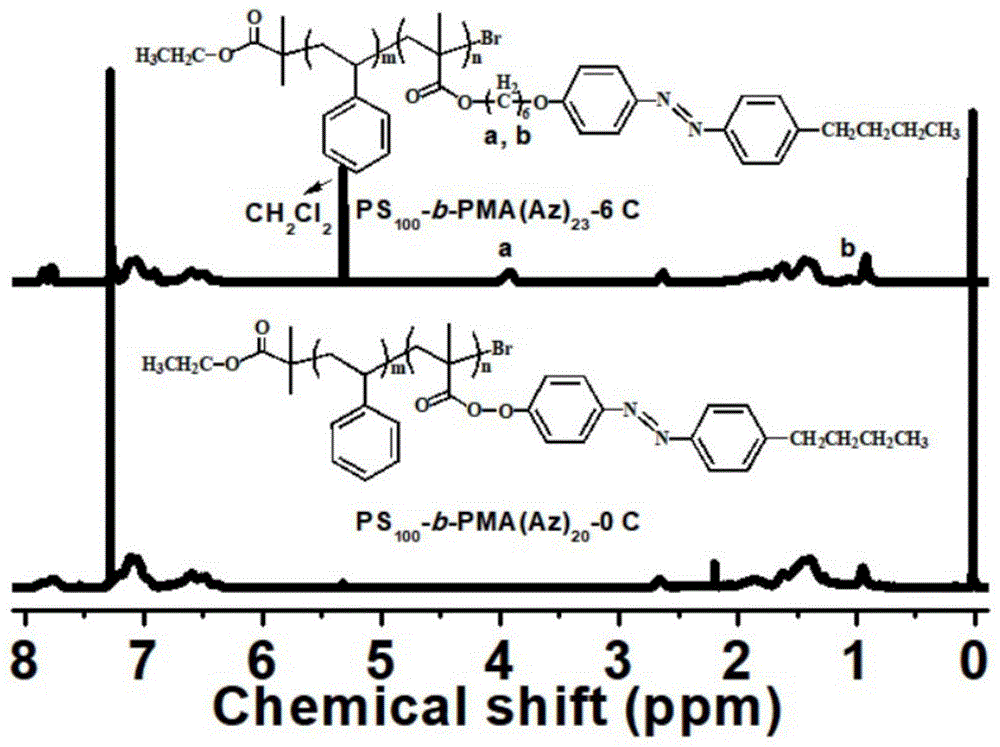
图1是合成的ps-b-pma(az)-0c和ps-b-pma(az)-6c两嵌段共聚物的1hnmr图,从上至下依次为ps100-b-pma(az)23-6c和ps100-b-pma(az)20-0c的1hnmr图;
图2是合成的ps,ps-b-pma(az)-11c两嵌段共聚物的1hnmr谱图,从上至下依次为ps28-b-pma(az)68-11c,ps100-b-pma(az)44-11c,ps42-b-pma(az)16-11c,ps60-b-pma(az)15-11c,ps100-b-pma(az)22-11c,ps100-b-pma(az)18-11c和ps的1hnmr谱图;
图3是合成的ps,ps-b-pma(az)-11c两嵌段共聚物的dsc降温曲线图,从下至上依次对应的是ps28,ps42,ps60,ps100,ps100-b-pma(az)18-11c,ps100-b-pma(az)22-11c,ps60-b-pma(az)15-11c,ps42-b-pma(az)16-11c,ps100-b-pma(az)44-11c和ps28-b-pma(az)68-11c的dsc曲线图;
图4是实施例6制备得到的ps-b-pma(az)-0c层状bcp薄膜rie后的断面sem图;
图5是实施例7制备得到的ps-b-pma(az)-6c层状bcp薄膜rie后的断面sem图;
图6是实施例8制备得到的ps-b-pma(az)-11c层状bcp薄膜rie后的上表面sem图;
图7是实施例8制备得到的ps-b-pma(az)-11c层状bcp薄膜rie后的断面sem图;
图8是实施例9制备得到的ps-b-pma(az)-11c柱状bcp薄膜rie后的上表面sem图;
图9是实施例9制备得到的ps-b-pma(az)-11c柱状bcp薄膜rie后的断面sem图;
图10是实施例10制备得到不同退火条件下、rie后薄膜的sem图,其中(a)为上表面sem图,(b)为断面sem图;
图11是实施例11制备得到的在不同支撑体上的退火和进一步rie后bcp薄膜的光学图和上表面sem图,其中,支撑体为铝箔所形成的bcp薄膜的光学图为图11中的(a),上表面sem图为图11中的(c)和(e);支撑体为pet所形成的bcp薄膜的光学图为图11中的(b),上表面sem图为图11中的(d)和(f)。
图12是实施例12制备得到的不同rie条件后bcp薄膜的上表面sem图和断面sem图,其中rie条件为:纯o2气体、体积流量为50sccm、刻蚀功率为50w、刻蚀时间为30s后得到的bcp薄膜,上表面sem图为图12中的(a)、断面sem图为图12中的(b);rie条件为o2和ar混合气体、体积流量为40/10sccm、刻蚀功率为50w、刻蚀时间为60s后得到的bcp薄膜,上表面sem图为图12中的(c)、断面sem图为图12中的(d);
图13是实施例13制备得到的不同柱状ps-b-pma(az)-11crie后薄膜的上表面sem图和断面sem图,其中,ps60-b-pma(az)15-11crie后薄膜上表面sem图为图13中的(a),断面sem图为图13中的(b);ps42-b-pma(az)16-11crie后薄膜上表面sem图为图13中的(c),断面sem图为图13中的(d);ps28-b-pma(az)68-11crie后薄膜上表面sem图为图13中的(e),断面sem图为图13中的(f)。
图14是实施例14制备得到的ps100-b-pma(az)18-11c层状bcp薄膜rie后的上表面sem图(a)和断面sem图(b)。
具体实施方式
以下实施例用于说明本发明,但不用来限制本发明的范围,以下实施例中涉及的操作如无特殊说明均为本领域常规操作。
实施例1
ps100-b-pma(az)20-0c嵌段共聚物的制备方法,包括如下步骤:
1)ps大分子引发剂的合成:
向干净的史莱克瓶中依次加入6ml除水后的氯苯,110倍α-溴代异丁酸乙酯摩尔当量的st即6.4ml,75.2ul的α-溴代异丁酸乙酯,125ul的配体五甲基二乙烯三胺(pmdeta),混合均匀。然后,将史莱克瓶置于液氮中冷冻,加入51mg氯化亚铜(cucl),再用胶塞将瓶上口封死。其中加料摩尔比为,引发剂:配体:cucl=1:1.2:1。随后在冷冻情况下抽真空5分钟,通氮气情况下解冻并搅拌5分钟,循环3次。最后,在冷冻抽真空的情况下将史莱克瓶的高真空截阀拧紧,再解冻并置于110℃油浴中搅拌反应16小时。
达到反应时间且体系黏度上升时,将体系用液氮淬灭,打开橡胶塞将得到的产物体系用二氯甲烷溶解并过中性氧化铝柱子除铜盐,用滴管吸取滤液并在高速搅拌的冷石油醚中沉淀和洗涤三次以除去未反应的st单体和低聚物。用砂芯漏斗将沉降后的悬浊抽滤,将抽滤得到的固体用少量二氯溶剂后,在高速搅拌的甲醇中沉淀和洗涤两次。再用砂芯漏斗将沉降后的悬浊抽滤,将抽滤得到的固体置于20℃真空烘箱中烘16h,最后得到纯净干燥的3.5g白色粉末,即嵌段数为100的ps100大分子引发剂;
2)ps100-b-pma(az)20-0c两嵌段共聚物的合成:
向干净的史莱克瓶中依次加入6ml除水后的氯苯,称0.5g的ps100大分子引发剂,0.4g的ma(az)-0c单体,12ul的配体五甲基二乙烯三胺(pmdeta),混合均匀。然后,将史莱克瓶置于液氮中冷冻2min,再抽真空2min后,通入氮气并在氮气氛下加入4.8mg氯化亚铜(cucl),再用胶塞将瓶上口封死。其中加料摩尔比为,引发剂:配体:cucl=1:1.2:1。随后在冷冻情况下抽真空5分钟,通氮气情况下解冻并搅拌5分钟,循环3次。然后在冷冻抽真空的情况下将史莱克瓶的高真空截阀拧紧,再解冻并置于110℃油浴中搅拌反应16小时。
16h后将体系用液氮淬灭,打开橡胶塞将得到的产物体系用二氯溶解并过中性氧化铝柱子除铜盐,用滴管吸取过滤液并在高速搅拌的冷石油醚中沉淀和洗涤三次,以除去未反应的单体和低聚物。最后用砂芯漏斗将沉降后的悬浊液抽滤,将抽滤得到的固体用少量二氯溶剂溶解后,再在高速搅拌的甲醇中沉淀和洗涤两次。最后用砂芯漏斗将沉降后的悬浊抽滤,将抽滤得到的固体置于20℃真空烘箱中烘16h。最后得到纯净干燥的0.6g淡黄色粉末,即嵌段数ps100-b-pma(az)20-0c的嵌段共聚物,其1hnmr图见图1。
实施例2
ps100-b-pma(az)23-6c嵌段共聚物的制备方法,同实施例1,其区别仅在于:
步骤1)中,以1-氯苯基乙烷(pe-cl)为引发剂,且引发剂:配体:cucl=1:2:1(摩尔比)。各物质的用量分别为:引发剂0.1g,配体297μl,cucl71mg,st9.0ml,反应溶剂氯苯6ml。最后得到嵌段数为100的ps100大分子引发剂6.1g;
步骤2)中,以ma(az)-6c为单体,三(2-二甲氨基乙基)胺(me6tren)为配体,且大分子引发剂:单体:配体:cucl=1:35:2:1(摩尔比),各物质的用量分别为:引发剂0.5g,单体ma(az)-6c为0.72g,配体14.4μl,cucl4.8mg;反应溶剂氯苯4ml,dmf2ml。ps100-b-pma(az)23-6c的1hnmr图见图1。
实施例3
ps42-b-pma(az)16-11c嵌段共聚物的制备方法,同实施例1,其区别仅在于:
步骤1)中,以α-溴代苯乙烷为引发剂,且引发剂:配体:cucl=1:1.5:1(摩尔比)。各原料的用量分别为:引发剂0.1g,配体121μl,cucl53.5mg;单体st2.8g,溶剂为3mldmf,3ml氯苯。最后得到嵌段数为42的ps42大分子引发剂2.0g。
步骤2)中,单体为ma(az)-11c,大分子引发剂:配体:cucl=1:1.5:1(摩尔比),各原料的用量分别为:引发剂0.5g,配体36μl,cucl11.4mg,单体ma(az)1.5g,溶剂为3ml四氢呋喃,3ml氯苯。最后得到0.8g聚合物。ps42-b-pma(az)16-11c的1hnmr图见图2,其dsc曲线图见图3。
实施例4
ps100-b-pma(az)22-11c嵌段共聚物的制备方法,同实施例1,其区别仅在于:
步骤1)中,以α-溴代异丁酸甲酯为引发剂,且引发剂:配体:cucl=1:2:1(摩尔比)。各原料的用量分别为:引发剂0.1g,配体231μl,cucl55mg,st6.4g,溶剂dmf3ml,氯苯3ml。最后得到嵌段数为100的ps100大分子引发剂5g;
步骤2)中,以五甲基二丙烯三胺为配体,且大分子引发剂:配体:cucl=1:2:1(摩尔比),各原料的用量分别为:引发剂0.5g,配体0.020g,cucl4.8mg;溶剂为氯苯6ml,单体ma(az)0.60g,即25摩尔当量的ps100的量。ps100-b-pma(az)22-11c的1hnmr图见图2,其dsc曲线图见图3。
实施例5
ps100-b-pma(az)44-11c嵌段共聚物的制备方法,同实施例1,其区别仅在于:
步骤1)中,引发剂:配体:cucl=1:1.5:1(摩尔比),各原料的用量分别为:引发剂0.1g,配体0.14g,cucl51mg,st5.86g,溶剂为氯苯4ml,dmf2ml,最后得到嵌段数为100的ps大分子引发剂3.2g。
步骤2)中,大分子引发剂:配体:cucl=1:1.5:1(摩尔比),各原料的用量分别为:引发剂0.5g,配体12.5mg,cucl4.8mg。单体ma(az)-11c为1.2g,溶剂为四氢呋喃2ml,氯苯2ml,dmf2ml。ps100-b-pma(az)44-11c的1hnmr谱图见图2,其dsc曲线图见图3。
实施例6
该实施例提供了垂直层状ps-b-pma(az)两嵌段共聚物光刻图案薄膜的制备,包括如下步骤:
(1)配制嵌段共聚物溶液:
取0.01g实施例1的ps-b-pma(az)-0c,fps=0.49-0.70,并将其溶解在0.99g氯仿中,室温下搅拌2h,得到浓度为1-3wt%的bcp/chcl3溶液。
(2)处理硅片并涂膜:
取2cm*2cm的单晶硅片置于丙酮中超声清洗30min,再置于乙醇中超声清洗30min。取出硅片,用氮气吹干硅片表面溶剂待用。用1ml滴管吸取1-3wt%的bcp/chcl3溶液滴到干净硅片上,1000-5000rpm下旋涂成膜,并置于室温环境下6h后待用。
(3)薄膜热退火:
将上述薄膜置于温度为120-200度的真空烘箱中,抽真空,保温5-10min,再缓慢冷却至60度,取出样品。
(4)薄膜rie:
将上述薄膜置于rie-100的样品室内,刻蚀条件为:o2、ar混合气体,体积流量为40/10sccm,刻蚀功率为50w,刻蚀时间为2-30s。
实施例7
该实施例提供层状ps-b-pma(az)-6c两嵌段共聚物光刻图案薄膜的制备,制备方法同实施例6,区别仅在于:使用实施例2制备得到的ps-b-pma(az)-6c,fps=0.49-0.70,两嵌段共聚物0.01g,配制成浓度为1-3wt%的旋涂溶液,旋涂转速为1000-5000rpm。
实施例8
该实施例提供了垂直层状ps-b-pma(az)-11c两嵌段共聚物光刻图案薄膜的制备,制备方法同实施例6,区别仅在于:使用实施例4制备得到的ps-b-pma(az)-11c,fps=0.49-0.70,配制旋涂液的溶剂为氯仿、四氢呋喃和甲苯任何比例的混合液,退火温度为100-140度,退火时间为5-10min。
实施例9
该实施例提供了垂直柱状ps-b-pma(az)-11c两嵌段共聚物光刻图案薄膜的制备,制备方法同实施例6,区别仅在于:使用实施例5制备得到的ps-b-pma(az)-11c,fps=0.001-0.48,配制旋涂液的溶剂甲苯,退火温度为100-140度,退火时间为5-10min。
实施例10
该实施例提供了不同退火条件下的垂直柱状ps-b-pma(az)-11c两嵌段共聚物光刻图案薄膜的制备,制备方法同实施例9,区别仅在于:退火温度为100-140度,退火时间为1min。从图8可以看出,退火时间太短得到的薄膜有序度不够。
实施例11
该实施例提供垂直柱状ps-b-pma(az)-11c两嵌段共聚物光刻图案薄膜的制备,制备方法同实施例9,区别仅在于:旋涂支撑体为pet和铝箔中的任意一种。
实施例12
该实施例提供不同光刻条件下柱状ps-b-pma(az)-11c两嵌段共聚物光刻图案薄膜的制备,制备方法同实施例9,区别仅在于rie条件不同:(1)纯o2气体,体积流量为50sccm,刻蚀功率为50w,刻蚀时间为30s;(2)o2和ar混合气体,体积流量为40/10sccm,刻蚀功率为50w,刻蚀时间为60s。从图10中可以看出,采用两种不同rie条件,pma(az)和ps嵌段在这两种rie条件下刻蚀差异性不大,ps柱子图案不能很好的保留下来。
实施例13
该实施例提供不同直径垂直柱状ps-b-pma(az)-11c两嵌段共聚物光刻图案薄膜的制备,制备方法同实施例9,区别仅在于:使用ps60-b-pma(az)15-11c,ps42-b-pma(az)16-11c,ps28-b-pma(az)68-11c,fps=0.001-0.48,两嵌段共聚物0.01g,配制成浓度为1-3wt%旋涂溶液,旋涂转速为1000-5000rpm。从图11中可以看出,选择的三种bcp的ps体积分数为:0.001~0.48,得到柱直径d约12~19nm的三种ps柱状光刻薄膜。
实施例14
该实施例提供不同层间距的垂直层状ps-b-pma(az)-11c两嵌段共聚物光刻图案薄膜的制备,制备方法同实施例8,区别仅在于:使用ps100-b-pma(az)18-11c,fps=0.49-0.58,两嵌段共聚物0.01g,配制成浓度为1-3wt%旋涂溶液,旋涂转速为1000-5000rpm。从图12中可以看出,该bcp的体积分数为0.55,得到层间距w约20~28nm的ps层状光刻薄膜。
为了进一步说明本发明的效果,将退火好的薄膜于液氮中淬冷,5s后取出并用玻璃刀切断,喷金后即制得薄膜断面电镜样品可用于电镜检测,从图4,5,6,7,8,9,10,11,12,13,14可以看出,退火时间太短得到的薄膜有序度不够,在合适的退火条件下能得到高度有序的垂直六方柱状或层状排列的薄膜,并且通过选择不同嵌段的ps,得到直径可调的垂直六方柱状结构薄膜和层间距可调的垂直层状薄膜。
此外,对实施例9制备得到的在不同支撑体上的薄膜,其中对柔性pet支撑体上的薄膜经过多次弯折,仍能得到高度有序排列的六方柱状结构。
虽然,上文中已经用一般性说明、具体实施方式及试验,对本发明作了详尽的描述,但在本发明基础上,可以对之作一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。
- 还没有人留言评论。精彩留言会获得点赞!