一种芳基丙醛类化合物的连续制备方法与流程
本发明涉及一种芳基丙醛类化合物的连续制备方法。
背景技术:
芳基丙醛类化合物是构建药物分子的重要中间体。例如,于2001年美国fda批准上市,由艾尔建公司开发的治疗青光眼药物比马前列素(式1);2004年fda批准上市的由amgen公司(nps制药公司该产品的许可权受让人)生产的,用于治疗进行透析的慢性肾病(ckd)患者的继发性甲状旁腺功能亢进症和用于治疗甲状旁腺癌患者的高钙血症的药物盐酸西那卡塞(式2);由辉瑞公司开发的正处于临床阶段的新药efipladib(式3)和uk-447841(式4)都用到了芳基丙醛类化合物作为中间体,该四种药物所使用的中间体分别为(式1-1)、(式2-1)、(式3-1)和(式4-1)。
目前,芳基丙醛类化合物的合成方法中一类是以芳基丙醇为底物,进行选择性氧化反应制得芳基丙醛(journaloforganicchemistry,74(22),8510-8515;2009;pctint.appl.,2001081325,01nov2001;bioorganic&medicinalchemistryletters,26(7),1849-1853;2016),或是以芳基丙酸酯为底物,进行选择性还原反应制得芳基丙醛(europeanjournaloforganicchemistry,(20),4573-4577;2006;catalysiscommunications,50,25-28;2014)。但是,该类方法原料不易获得,氧化剂和还原剂的成本均较高,且反应过程中芳基丙醇易被氧化为芳基丙酸,芳基丙酸酯易被还原为芳基丙醇,反应不好控制,这类氧化还原反应都比较危险,不适合工业化规模的生产。另一类是以芳基卤化物或硼化物跟烯丙基醇或烯丙基醛发生heck和suzuki等偶联反应制得芳基丙醛。其中,heck反应是以卤代烃与烯基化合物在钯催化剂作用下形成碳碳键的偶联反应,被广泛应用在有机合成和制药领域(greenchemistry,16(5),2788-2797;2014;cn102120718b;bioorganic&medicinalchemistryletters,27(2),237-241;2017;journalofmedicinalchemistry,56(23),9427-9440;2013;organicletters,13(20),5456-5459;2011)。但heck反应对空气和水分非常敏感,反应时间往往比较长,反应效率较低,该方法目前都是采用比较传统的间歇式反应。因此,存在钯催化剂容易失活,催化剂用量较多,成本高,产品质量不稳定,也不利于工业化生产等问题。organicletters,13(20),5456-5459;2011中介绍以溴苯和烯丙醇为原料,pd(dba)2为催化剂,n,n-二环己基甲胺为有机碱,n,n-二甲基甲酰胺为溶剂,如下式5所示的化合物为配体,在100℃条件下反应1h,收率为74%。溴苯与pd(dba)2的摩尔比为1:0.02,溴苯与有机碱的摩尔比为1:1.1,溴苯与配体的摩尔比为1:0.06。此方法催化剂用量多,如下式5所示的配体价格高,反应时间长,且使用间歇式反应,不适用于工业化生产。
连续制备方法比较适用于大宗产品的生产,但是连续流动反应器比较适用于反应时间较短的反应或者在循环条件下产物稳定的反应。进行这个操作过程中需要的条件:必须在理想的温度、流速和其他关键参数的条件下,达到合适的反应完成程度,需要一定的停留时间。连续式操作也缺乏灵活性(安德森.实用有机合成工艺研发手册[m].科学出版社,2011.)。可见,并不是所有反应均可在连续反应器中进行。且连续式操作过程有许多缺陷。
因此,本领域亟需一种反应时间短、工艺易于控制、成本低、安全高效、催化剂用量少且保证催化剂在反应过程具有较高活性的方法,用来解决上述技术上难题。
技术实现要素:
本发明所要解决的技术问题是为了克服现有的芳基丙醛类化合物在制备过程中催化剂易失活且用量多,反应时间长,成本高,不适用于工业化生产等缺陷,而提供一种芳基丙醛类化合物的连续制备方法。本发明的制备方法缩短反应时间,降低催化剂用量,安全性好,保证产品质量,降低成本,减少副产物焦油的生成,更适用于工业化生产。
现有技术中通过heck反应制备芳基丙醛类化合物的方法的反应时间普遍比较长,且在高温和长时间反应的条件下,易产生焦油;而连续制备方法又比较适用于反应时间比较短的化学反应,或是循环条件下产物稳定的反应。本发明在管式反应器中进行heck反应,通过创造性的实验克服了上述技术偏见,缩短反应时间,减少催化剂和配体用量,使用常规配体,降低反应成本。
本发明主要是通过以下技术方案解决以上技术问题。
本发明提供一种芳基丙醛类化合物的连续制备方法,其包括下列步骤:溶剂中,在钯催化剂、配体和有机碱的作用下,将式a所示的化合物与式b所示的化合物在管式反应器中进行如下所示的反应,制得式c所示的化合物;
其中,x为cl、br、i或三氟甲磺酸酯基;
r为氢、c1-c6烷基或氰基
r1、r2、r3、r4和r5各自独立地为h、f、cl、卤素取代c1-c4烷基、c1-c4烷基、卤素取代c1-c4烷氧基、c1-c4烷氧基、c2-c5杂芳基、c6-c10芳基、硝基
本发明中,所述的卤素优选cl、br或i。
本发明中,所述的c1-c4烷基优选甲基、乙基、正丙基、异丙基、正丁基、异丁基或叔丁基。
本发明中,所述的卤素取代c1-c4烷基是指被一个或多个卤素取代的c1-c4烷基,本发明优选三氟甲基。
本发明中,所述的卤素取代c1-c4烷氧基是指被一个或多个卤素取代的c1-c4烷氧基,本发明优选三氟甲氧基。
本发明中,所述的c1-c4烷氧基优选甲氧基。
本发明中,所述的c2-c5杂芳基优选吡啶基、噻吩基、噻唑基、哌啶基或哌嗪基。
本发明中,所述c6-c10芳基优选苯基。
所述的钯催化剂可为本领域中此类反应常规的钯催化剂,本发明优选pd2(dba)3、pd(oac)2、pd(pph3)4、pd(dba)2和pdcl2中的一种或几种,进一步优选pd2(dba)3。
所述的有机碱可为本领域中此类反应常规的有机碱,本发明优选胺类有机化合物和/或季铵盐,进一步优选n,n-二环己基甲胺、三乙胺、三丁基胺、二异丙基乙胺、四丁基溴化铵、四丁基氯化铵、吡啶和联吡啶(例如,2,2`-联吡啶、2,3`-联吡啶和4,4`-联吡啶中的一种或几种)中的一种或几种,更优选n,n-二环己基甲胺。
所述的配体可为本领域中此类反应常规的配体,本发明优选四氟硼酸三叔丁基膦、三苯基膦、三邻甲基苯基膦和三环己基膦中的一种或几种,进一步优选四氟硼酸三叔丁基膦。
所述的溶剂可为本领域中此类反应常规的溶剂,以不影响反应的进行,即可,本发明优选酰胺类溶剂、醚类溶剂、吡咯烷酮类溶剂和芳烃类溶剂中的一种或几种;所述酰胺类溶剂优选n,n-二甲基甲酰胺和/或n,n-二甲基乙酰胺,进一步优选n,n-二甲基甲酰胺;所述醚类溶剂优选1,4-二氧六环;所述吡咯烷酮类溶剂优选n-甲基吡咯烷酮;所述芳烃类溶剂优选甲苯和/或二甲苯(例如,邻二甲苯、间二甲苯和对二甲苯中的一种或几种)。
所述式a所示的化合物、所述钯催化剂、所述配体和所述有机碱的用量不做具体限定。本领域众所周知,本发明所述反应中所述催化剂、所述配体和所述有机碱用量增多,除增加成本不经济外,对反应的质量和收率影响不大,只要控制所述催化剂、所述配体和所述有机碱的用量下限值,即可。本发明所述式a所示的化合物与所述催化剂的摩尔比优选1:0.0001~1:0.015,进一步优选1:0.0003~1:0.01,更优选1:0.0004~1:0.008,例如1:0.004。
所述式a所示的化合物与所述有机碱的摩尔比优选1:0.5~1:4,进一步优选1:0.5~1:2,更优选1:0.9~1:1.5,例如1:1.01、1:1.1或1:1.21。
所述式a所示的化合物与所述配体的摩尔比优选1:0.0003~1:0.5,进一步优选1:0.0003~1:0.1,更优选1:0.0008~1:0.06,最优选1:0.001~1:0.03,例如1:0.0011、1:0.005或1:0.011。
所述式a所示的化合物与所述式b所示的化合物的摩尔比优选1:1~1:5,进一步优选1:1.17~1:4,更优选1:1.5~1:4,最优选1:1.7~1:3.4,例如1:2.5。
所述溶剂的用量可不做具体限定,以不影响反应的进行,即可;所述溶剂与所述式a所示的化合物的质量比优选1:1~10:1,进一步优选1:1~4:1,更优选2.5:1~3.1:1,例如2.9:1。
所述反应的反应时间可为0.1min~30min,优选0.1min~10min,进一步优选0.5min~8min,例如1min、2.9min、4.5min或5min。
所述反应的反应温度为本领域此类反应常规的温度,本发明优选10℃~200℃,进一步优选30℃~150℃,更优选110℃~140℃,最优选120℃~130℃。
由于管式反应器本身就能够提供无水无氧环境,故本发明的反应无需额外控制无水无氧条件。
在本发明一优选实施方案中,所述的r为h,所述的r1、r2、r3、r4和r5为氢,所述的x为cl、br或i,优选br。
当所述的r为h,所述的r1、r2、r3、r4和r5为氢,所述的x为cl、br或i时,所述的有机碱优选胺类有机化合物。所述式a所示的化合物与所述催化剂的摩尔比优选1:0.0003~1:0.01。所述式a所示的化合物与所述配体的摩尔比优选1:0.0008~1:0.06。所述式a所示的化合物与所述式b所示的化合物的摩尔比优选1:1.5~1:4。所述反应的反应时间优选0.1min~10min。所述反应的反应温度优选110℃~140℃。其余条件同前所述。
在本发明一优选实施方案中,所述的r为h,所述的r1、r2、r4和r5为氢,所述的r3为f或cl,优选cl,所述的x为cl、br或i,优选i。
当所述的r为h,所述的r1、r2、r4和r5为氢,所述的r3为f或cl,所述的x为cl、br或i时,所述的有机碱优选胺类有机化合物。所述式a所示的化合物与所述催化剂的摩尔比优选1:0.0003~1:0.01。所述式a所示的化合物与所述有机碱的摩尔比优选1:0.9~1:1.5。所述式a所示的化合物与所述配体的摩尔比优选1:0.0008~1:0.06。所述式a所示的化合物与所述式b所示的化合物的摩尔比优选1:1.5~1:4。所述溶剂与所述式a所示的化合物的质量比优选1:1~4:1。所述反应的反应时间优选0.1min~10min。所述反应的反应温度优选110℃~140℃。其余条件同前所述。
在本发明一优选实施方案中,所述的r为h,所述的r1、r2、r4和r5为氢,所述的r3为所述的
当所述的r为h,所述的r1、r2、r4和r5为氢,所述的r3为所述的
所述芳基丙醛类化合物的连续制备方法优选包括下列步骤:
(1)将所述式a所示的化合物、所述钯催化剂、所述有机碱、所述配体和所述溶剂形成混合液1;
(2)当所述式b所示的化合物为固体时,将所述式b所示的化合物与所述溶剂形成混合液2;将所述混合液1与所述混合液2分别经计量泵进入管式反应器,进行所述反应;所述混合液2的浓度不做具体限定,满足所述混合液2不堵塞所述管式反应器的管路,即可。
当所述式b所示的化合物为液体时,将所述混合液1和所述式b所示的化合物分别经计量泵进入管式反应器,进行所述反应。
所述混合液1的流速在所述管式反应器本身的流速允许范围之内,可为本领域采用所述管式反应器进行此类反应常规所用的流速,优选0.01ml/min~50l/min,进一步优选1ml/min~10l/min,更优选30ml/min~50ml/min,例如32.9ml/min、34.5ml/min、36.8ml/min、37.6ml/min、40ml/min、41.4ml/min或50ml/min。
当所述式b所示的化合物为固体时,所述混合液2的流速在所述管式反应器本身的流速允许范围之内,可为本领域采用所述管式反应器进行此类反应常规所用的流速,优选0.01ml/min~50l/min,进一步优选1ml/min~10l/min,更优选2ml/min~15ml/min,最优选3.6ml/min~10.5ml/min,例如5.2ml/min、5.7ml/min或7ml/min。
当所述式b所示的化合物为液体时,所述式b所示的化合物的流速在所述管式反应器本身的流速允许范围之内,可为本领域采用所述管式反应器进行此类反应常规所用的流速,优选0.01ml/min~50l/min,进一步优选1ml/min~10l/min,更优选2ml/min~15ml/min,最优选3.6ml/min~10.5ml/min,例如5.2ml/min、5.7ml/min或7ml/min。
当所述的r为h,所述的r1、r2、r3、r4和r5为氢,所述的x为cl、br或i时,所述混合液1的流速优选30ml/min~50ml/min。所述式b所示的化合物的流速优选2ml/min~15ml/min。
当所述的r为h,所述的r1、r2、r4和r5为氢,所述的r3为f或cl,所述的x为cl、br或i时,所述混合液1的流速优选30ml/min~50ml/min。所述式b所示的化合物的流速优选2ml/min~15ml/min。
当所述的r为h,所述的r1、r2、r4和r5为氢,所述的r3为所述的
本发明中,所述的管式反应器优选带有t型混合器的管式反应器,进一步优选申请号为201720662235.6的专利公开的管式反应装置。
所述芳基丙醛类化合物制备结束后还进一步包括后处理操作。所述的后处理过程可采用本领域此类反应的常规后处理操作,本发明优选包括如下步骤:将反应结束后得到粗产物从所述带有t型混合器的管式反应器连续流出至接收器,降温至0~60℃,加入乙酸乙酯,再加入水洗涤有机层,蒸出溶剂,经过蒸馏或重结晶得到芳基丙醛类化合物。
本发明中,时间描述中的字母min是指分钟。
在不违背本领域常识的基础上,上述各优选条件可任意组合,即得本发明各较佳实例。
本发明所用试剂和原料均市售可得。
本发明的积极进步效果在于:1、该反应工艺为连续流反应,与间歇反应相比,反应时间缩短,可提高反应温度,克服间歇反应在温度升高时生成焦油的困难,该反应工艺提高产率;2、原料持续加入带有t型混合器的管式反应器中,反应液持续流出至接收器,避免了间歇反应中热量积累给生产带来的安全隐患;3、与间歇反应相比,避免发生催化剂长时间在高温下失活的现象,提高催化剂的活性,该工艺催化剂用量减少至现有技术的1/50~1/5;4、该工艺减少了副产物焦油的生成,分离纯化步骤简单,适用于工业化生产;5、该工艺克服了反应时间普遍较长的heck反应不能在管式反应器中进行反应的技术偏见。
附图说明
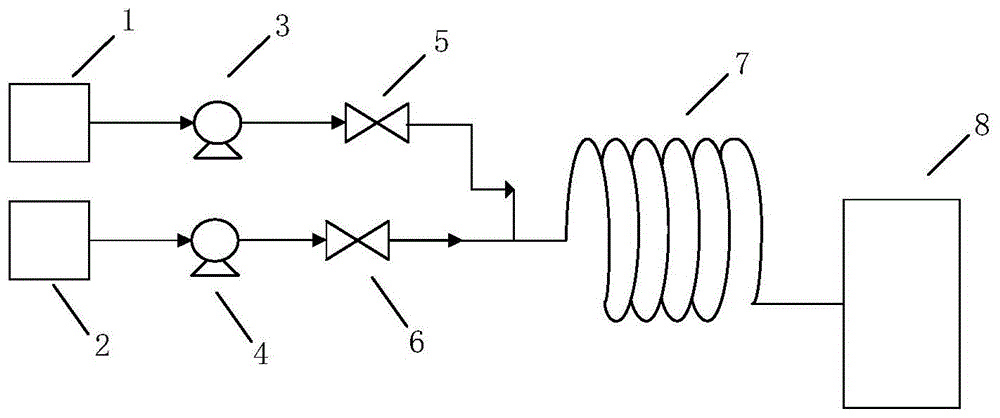
图1为本发明所使用的带有t型混合器的管式反应器的工艺流程示意图。
其中,1和2表示原料罐,3和4表示计量泵,5和6表示截止阀,7表示带有t型混合器的管式反应器,8表示收集罐。
具体实施方式
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
实施例1
1、原料配置:称取溴苯(70g,0.445mol)溶解在n,n-二甲基甲酰胺溶剂(206g)中,加入pd2(dba)3(1.7g,0.0018mol,0.004eq),四氟硼酸三叔丁基膦(1.4g,0.0049mol,0.011eq)和n,n-二环己基甲胺(87.6g,0.45mol,1.01eq),搅拌使其混合均匀制得混合液1,混合液1的密度为1.0g/ml,将混合液1放于原料罐1;称取烯丙醇(44.4g,0.76mol),烯丙醇密度为0.85g/ml,放于原料罐2中;
2、利用本发明的装置图1,按照下述步骤:(1)将原料罐1中混合液1通过计量泵3进入带有t型混合器的管式反应器7,原料罐2中的烯丙醇通过计量泵4进入带有t型混合器的管式反应器7;(2)设定计量泵3和计量泵4控制混合液1和烯丙醇的流速分别为40ml/min和5.7ml/min,设定带有t型混合器的管式反应器7的温度为120℃,反应停留时间2.9min;(3)反应通道上通过截止阀5和截止阀6来防止物料的倒流;(4)在经过带有t型混合器的管式反应器7混合反应后,粗产物连续出料收集到收集罐8中,粗产物经过hplc分析;(4)粗产物降温至25℃,粗产物中加入乙酸乙酯(700g),过滤除去生成的盐和所述钯催化剂,加入水(200g*3)洗涤有机层,蒸出溶剂,经过蒸馏或重结晶,产物经过gc-ms分析。
3、溴苯转化率为99.3%(hplc分析结果);粗产物纯化后,产物纯度为97.5%,收率为82%(gc-ms分析结果)。
实施例2
1、原料配置:称取溴苯(70g,0.445mol)溶解在n,n-二甲基甲酰胺溶剂(206g)中,加入pd2(dba)3(0.17g,0.18mmol,0.0004eq),四氟硼酸三叔丁基膦(0.14g,0.49mmol,0.0011eq)和n,n-二环己基甲胺(87.6g,0.45mol,1.01eq),搅拌使其混合均匀制得混合液1,混合液1的密度为1.0g/ml,将混合液1放于原料罐1;称取烯丙醇(44.4g,0.76mol),烯丙醇密度为0.85g/ml,放于原料罐2中;
2、利用本发明的装置图1,按照下述步骤:(1)将原料罐1中混合液1通过计量泵3进入带有t型混合器的管式反应器7,原料罐2中的烯丙醇通过计量泵4进入带有t型混合器的管式反应器7;(2)设定计量泵3和计量泵4控制混合液1和烯丙醇的流速分别为40ml/min和5.7ml/min,设定带有t型混合器的管式反应器7的温度为130℃,反应停留时间4.5min;(3)反应通道上通过截止阀5和截止阀6来防止物料的倒流;(4)在经过带有t型混合器的管式反应器7混合反应后,粗产物连续出料收集到收集罐8中,粗产物经过hplc分析;(4)粗产物降温至25℃,粗产物中加入乙酸乙酯(700g),过滤除去生成的盐和所述钯催化剂,加入水(200g*3)洗涤有机层,蒸出溶剂,经过蒸馏或重结晶,产物经过gc-ms分析。
3、溴苯转化率为96.5%(hplc分析结果);粗产物纯化后,产物纯度为97.1%,收率为78%(gc-ms分析结果)。
实施例3
1、原料配置:称取溴苯(70g,0.445mol)溶解在n,n-二甲基甲酰胺溶剂(206g)中,加入pd(oac)2(1g,4.45mmol,0.01eq),三苯基膦(0.58g,2.21mmol,0.005eq)和三丁基胺(90.73g,0.49mol,1.1eq),搅拌使其混合均匀制得混合液1,混合液1的密度为1.0g/ml,将混合液1放于原料罐1;称取烯丙醇(44.4g,0.76mol),烯丙醇密度为0.85g/ml,放于原料罐2中;
2、利用本发明的装置图1,按照下述步骤:(1)将原料罐1中混合液1通过计量泵3进入带有t型混合器的管式反应器7,原料罐2中的烯丙醇通过计量泵4进入带有t型混合器的管式反应器7;(2)设定计量泵3和计量泵4控制混合液1和烯丙醇的流速分别为36.8ml/min和5.2ml/min,设定带有t型混合器的管式反应器7的温度为130℃,反应停留时间5min;(3)反应通道上通过截止阀5和截止阀6来防止物料的倒流;(4)在经过带有t型混合器的管式反应器7混合反应后,粗产物连续出料收集到收集罐8中,粗产物经过hplc分析;(4)粗产物降温至25℃,粗产物中加入乙酸乙酯(700g),过滤除去生成的盐和所述钯催化剂,加入水(200g*3)洗涤有机层,蒸出溶剂,经过蒸馏或重结晶,产物经过gc-ms分析。
3、溴苯转化率为92%(hplc分析结果);粗产物纯化后,产物纯度为97.1%,收率为78.5%(gc-ms分析结果)。
实施例4
1、原料配置:称取溴苯(70g,0.445mol)溶解在n,n-二甲基甲酰胺溶剂(206g)中,加入pd2(dba)3(4.07g,4.45mmol,0.01eq),四氟硼酸三叔丁基膦(0.65g,2.24mmol,0.005eq)和n,n-二环己基甲胺(95.72g,0.49mol,1.1eq),搅拌使其混合均匀制得混合液1,混合液1的密度为1.0g/ml,将混合液1放于原料罐1;称取烯丙醇(44.4g,0.76mol),烯丙醇密度为0.85g/ml,放于原料罐2中;
2、利用本发明的装置图1,按照下述步骤:(1)将原料罐1中混合液1通过计量泵3进入带有t型混合器的管式反应器7,原料罐2中的烯丙醇通过计量泵4进入带有t型混合器的管式反应器7;(2)设定计量泵3和计量泵4控制混合液1和烯丙醇的流速分别为37.6ml/min和5.2ml/min,设定带有t型混合器的管式反应器7的温度为130℃,反应停留时间5min;(3)反应通道上通过截止阀5和截止阀6来防止物料的倒流;(4)在经过带有t型混合器的管式反应器7混合反应后,粗产物连续出料收集到收集罐8中,粗产物经过hplc分析;(4)粗产物降温至25℃,粗产物中加入乙酸乙酯(700g),过滤除去生成的盐和所述钯催化剂,加入水(200g*3)洗涤有机层,蒸出溶剂,经过蒸馏或重结晶,产物经过gc-ms分析。
3、溴苯转化率为100%(hplc分析结果);粗产物纯化后,产物纯度为98.2%,收率为82.1%(gc-ms分析结果)。
实施例5
1、原料配置:称取溴苯(70g,0.445mol)溶解在n,n-二甲基甲酰胺溶剂(206g)中,加入pd(dba)2(2.56g,4.45mmol,0.01eq),三邻甲基苯基膦(0.68g,2.23mmol,0.005eq)和三乙胺(49.58g,0.49mol,1.1eq),搅拌使其混合均匀制得混合液1,混合液1的密度为1.0g/ml,将混合液1放于原料罐1;称取烯丙醇(44.4g,0.76mol),烯丙醇密度为0.85g/ml,放于原料罐2中;
2、利用本发明的装置图1,按照下述步骤:(1)将原料罐1中混合液1通过计量泵3进入带有t型混合器的管式反应器7,原料罐2中的烯丙醇通过计量泵4进入带有t型混合器的管式反应器7;(2)设定计量泵3和计量泵4控制混合液1和烯丙醇的流速分别为32.9ml/min和5.2ml/min,设定带有t型混合器的管式反应器7的温度为130℃,反应停留时间5min;(3)反应通道上通过截止阀5和截止阀6来防止物料的倒流;(4)在经过带有t型混合器的管式反应器7混合反应后,粗产物连续出料收集到收集罐8中,粗产物经过hplc分析;(4)粗产物降温至25℃,粗产物中加入乙酸乙酯(700g),过滤除去生成的盐和所述钯催化剂,加入水(200g*3)洗涤有机层,蒸出溶剂,经过蒸馏或重结晶,产物经过gc-ms分析。
3、溴苯转化率为88%(hplc分析结果);粗产物纯化后,产物纯度为97.1%,收率为72.2%(gc-ms分析结果)。
实施例6
1、原料配置:称取溴苯(70g,0.445mol)溶解在n,n-二甲基甲酰胺溶剂(206g)中,加入pd(pph3)4(5.14g,4.45mmol,0.01eq),三环己基磷(0.63g,2.25mmol,0.005eq)和二异丙基乙胺(63.32g,0.49mol,1.1eq),搅拌使其混合均匀制得混合液1,混合液1的密度为1.0g/ml,将混合液1放于原料罐1;称取烯丙醇(88.8g,1.52mol),烯丙醇密度为0.85g/ml,放于原料罐2中;
2、利用本发明的装置图1,按照下述步骤:(1)将原料罐1中混合液1通过计量泵3进入带有t型混合器的管式反应器7,原料罐2中的烯丙醇通过计量泵4进入带有t型混合器的管式反应器7;(2)设定计量泵3和计量泵4控制混合液1和烯丙醇的流速分别为34.5ml/min和10.5ml/min,设定带有t型混合器的管式反应器7的温度为130℃,反应停留时间5min;(3)反应通道上通过截止阀5和截止阀6来防止物料的倒流;(4)在经过带有t型混合器的管式反应器7混合反应后,粗产物连续出料收集到收集罐8中,粗产物经过hplc分析;(4)粗产物降温至25℃,粗产物中加入乙酸乙酯(700g),过滤除去生成的盐和所述钯催化剂,加入水(200g*3)洗涤有机层,蒸出溶剂,经过蒸馏或重结晶,产物经过gc-ms分析。
3、溴苯转化率为81%(hplc分析结果);粗产物纯化后,产物纯度为96.8%,收率为60.4%(gc-ms分析结果)。
实施例7
1、原料配置:称取溴苯(70g,0.445mol)溶解在n,n-二甲基甲酰胺溶剂(206g)中,加入pd2(dba)3(1.7g,0.0018mol,0.004eq),四氟硼酸三叔丁基膦(1.4g,0.0049mol,0.011eq)和n,n-二环己基甲胺(87.6g,0.45mol,1.01eq),搅拌使其混合均匀制得混合液1,混合液1的密度为1.0g/ml,将混合液1放于原料罐1;称取烯丙醇(44.4g,0.76mol),烯丙醇密度为0.85g/ml,放于原料罐2中;
2、利用本发明的装置图1,按照下述步骤:(1)将原料罐1中混合液1通过计量泵3进入带有t型混合器的管式反应器7,原料罐2中的烯丙醇通过计量泵4进入带有t型混合器的管式反应器7;(2)设定计量泵3和计量泵4控制混合液1和烯丙醇的流速分别为40ml/min和5.7ml/min,设定带有t型混合器的管式反应器7的温度为130℃,反应停留时间0.5min;(3)反应通道上通过截止阀5和截止阀6来防止物料的倒流;(4)在经过带有t型混合器的管式反应器7混合反应后,粗产物连续出料收集到收集罐8中,粗产物经过hplc分析;(4)粗产物降温至25℃,粗产物中加入乙酸乙酯(700g),过滤除去生成的盐和所述钯催化剂,加入水(200g*3)洗涤有机层,蒸出溶剂,经过蒸馏或重结晶,产物经过gc-ms分析。
3、溴苯转化率为98.2%(hplc分析结果);粗产物纯化后,产物纯度为97.6%,收率为75%(gc-ms分析结果)。
实施例8
1、原料配置:称取溴苯(70g,0.445mol)溶解在n,n-二甲基甲酰胺溶剂(206g)中,加入pdcl2(0.79g,4.45mmol,0.01eq),三环己基磷(0.63g,2.25mmol,0.005eq)和四丁基氯化铵(136.18g,0.49mol,1.1eq),搅拌使其混合均匀制得混合液1,混合液1的密度为1.0g/ml,将混合液1放于原料罐1;称取烯丙醇(30.4g,0.52mol),烯丙醇密度为0.85g/ml,放于原料罐2中;)
2、利用本发明的装置图1,按照下述步骤:(1)将原料罐1中混合液1通过计量泵3进入带有t型混合器的管式反应器7,原料罐2中的烯丙醇通过计量泵4进入带有t型混合器的管式反应器7;(2)设定计量泵3和计量泵4控制混合液1和烯丙醇的流速分别为41.4ml/min和3.6ml/min,设定带有t型混合器的管式反应器7的温度为130℃,反应停留时间5min;(3)反应通道上通过截止阀5和截止阀6来防止物料的倒流;(4)在经过带有t型混合器的管式反应器7混合反应后,粗产物连续出料收集到收集罐8中,粗产物经过hplc分析;(4)粗产物降温至25℃,粗产物中加入乙酸乙酯(700g),过滤除去生成的盐和所述钯催化剂,加入水(200g*3)洗涤有机层,蒸出溶剂,经过蒸馏或重结晶,产物经过gc-ms分析。
3、溴苯转化率为71%(hplc分析结果);粗产物纯化后,产物纯度为96.5%,收率为43.6%(gc-ms分析结果)。
实施例9
1、原料配置:称取溴苯(70g,0.445mol)溶解在n,n-二甲基甲酰胺溶剂(206g)中,加入pd2(dba)3(1.7g,0.0018mol,0.004eq),四氟硼酸三叔丁基膦(1.4g,0.0049mol,0.011eq)和n,n-二环己基甲胺(87.6g,0.45mol,1.01eq),搅拌使其混合均匀制得混合液1,混合液1的密度为1.0g/ml,将混合液1放于原料罐1;称取烯丙醇(44.4g,0.76mol),烯丙醇密度为0.85g/ml,放于原料罐2中;
2、利用本发明的装置图1,按照下述步骤:(1)将原料罐1中混合液1通过计量泵3进入带有t型混合器的管式反应器7,原料罐2中的烯丙醇通过计量泵4进入带有t型混合器的管式反应器7;(2)设定计量泵3和计量泵4控制混合液1和烯丙醇的流速分别为40ml/min和5.7ml/min,设定带有t型混合器的管式反应器7的温度为200℃,反应停留时间8min;(3)反应通道上通过截止阀5和截止阀6来防止物料的倒流;(4)在经过带有t型混合器的管式反应器7混合反应后,粗产物连续出料收集到收集罐8中,粗产物经过hplc分析;(4)粗产物降温至25℃,粗产物中加入乙酸乙酯(700g),过滤除去生成的盐和所述钯催化剂,加入水(200g*3)洗涤有机层,蒸出溶剂,经过蒸馏或重结晶,产物经过gc-ms分析。
3、溴苯转化率为99.8%(hplc分析结果);粗产物纯化后,产物纯度为96.5%,收率为41%(gc-ms分析结果)。
实施例10
1、原料配置:称取溴苯(70g,0.445mol)溶解在n,n-二甲基甲酰胺溶剂(206g)中,加入pd2(dba)3(0.085g,0.09mmol,0.0002eq),四氟硼酸三叔丁基膦(0.07g,0.25mmol,0.00056eq)和n,n-二环己基甲胺(87.6g,0.45mol,1.01eq),搅拌使其混合均匀制得混合液1,混合液1的密度为1.0g/ml,将混合液1放于原料罐1;称取烯丙醇(44.4g,0.76mol),烯丙醇密度为0.85g/ml,放于原料罐2中;
2、利用本发明的装置图1,按照下述步骤:(1)将原料罐1中混合液1通过计量泵3进入带有t型混合器的管式反应器7,原料罐2中的烯丙醇通过计量泵4进入带有t型混合器的管式反应器7;(2)设定计量泵3和计量泵4控制混合液1和烯丙醇的流速分别为40ml/min和5.7ml/min,设定带有t型混合器的管式反应器7的温度为130℃,反应停留时间1min;(3)反应通道上通过截止阀5和截止阀6来防止物料的倒流;(4)在经过带有t型混合器的管式反应器7混合反应后,粗产物连续出料收集到收集罐8中,粗产物经过hplc分析;(4)粗产物降温至25℃,粗产物中加入乙酸乙酯(700g),过滤除去生成的盐和所述钯催化剂,加入水(200g*3)洗涤有机层,蒸出溶剂,经过蒸馏或重结晶,产物经过gc-ms分析。
3、溴苯转化率为43.9%(hplc分析结果);粗产物纯化后,产物纯度为98.2%,收率为28%(gc-ms分析结果)。
实施例11
1、原料配置:称取溴苯(70g,0.445mol)溶解在n,n-二甲基甲酰胺溶剂(206g)中,加入pd2(dba)3(1.7g,0.0018mol,0.004eq),四氟硼酸三叔丁基膦(1.4g,0.005mol,0.011eq)和n,n-二环己基甲胺(87.6g,0.45mol,1.01eq),搅拌使其混合均匀制得混合液1,混合液1的密度为1.0g/ml,将混合液1放于原料罐1;称取烯丙醇(44.4g,0.76mol),烯丙醇密度为0.85g/ml,放于原料罐2中;
2、利用本发明的装置图1,按照下述步骤:(1)将原料罐1中混合液1通过计量泵3进入带有t型混合器的管式反应器7,原料罐2中的烯丙醇通过计量泵4进入带有t型混合器的管式反应器7;(2)设定计量泵3和计量泵4控制混合液1和烯丙醇的流速分别为40ml/min和5.7ml/min,设定带有t型混合器的管式反应器7的温度为30℃,反应停留时间2.9min;(3)反应通道上通过截止阀5和截止阀6来防止物料的倒流;(4)在经过带有t型混合器的管式反应器7混合反应后,粗产物连续出料收集到收集罐8中,粗产物经过hplc分析;(4)粗产物降温至25℃,粗产物中加入乙酸乙酯(700g),过滤除去生成的盐和所述钯催化剂,加入水(200g*3)洗涤有机层,蒸出溶剂,经过蒸馏或重结晶,产物经过gc-ms分析。
3、溴苯转化率为9.8%(hplc分析结果);粗产物纯化后,产物纯度为98.5%,收率为5%(gc-ms分析结果)。
实施例12
1、原料配置:称取3-氯-碘苯(81g,0.34mol)溶解在n,n-二甲基甲酰胺溶剂(240g)中,加入pd2(dba)3(2.6g,0.0028mol,0.008eq),四氟硼酸三叔丁基膦(3.0g,0.01mol,0.03eq)和n,n-二环己基甲胺(80g,0.41mol,1.21eq),搅拌使其混合均匀制得混合液1,混合液1的密度为1.0g/ml,将混合液1放于原料罐1;称取烯丙醇(49g,0.84mol),烯丙醇密度为0.85g/ml,放于原料罐2中;
2、利用本发明的装置图1,按照下述步骤:(1)将原料罐1中混合液1通过计量泵3进入带有t型混合器的管式反应器7,原料罐2中的烯丙醇通过计量泵4进入带有t型混合器的管式反应器7;(2)设定计量泵3和计量泵4控制混合液1和烯丙醇的流速分别为50ml/min和7.0ml/min,设定带有t型混合器的管式反应器7的温度为130℃,反应停留时间5min;(3)反应通道上通过截止阀5和截止阀6来防止物料的倒流;(4)在经过带有t型混合器的管式反应器7混合反应后,粗产物连续出料收集到收集罐8中,粗产物经过hplc分析;(4)粗产物降温至25℃,粗产物中加入乙酸乙酯(700g),过滤除去生成的盐和所述钯催化剂,加入水(200g*3)洗涤有机层,蒸出溶剂,经过蒸馏或重结晶,产物经过gc-ms分析。
3、3-氯-碘苯转化率为99.5%(hplc分析结果);粗产物纯化后,产物纯度为98.2%,收率为69%(gc-ms分析结果)。
实施例13
1、原料配置:称取4-溴-苯甲酸乙酯(78g,0.34mol)溶解在n,n-二甲基甲酰胺溶剂(240g)中,加入pd2(dba)3(2.6g,0.0028mol,0.008eq),四氟硼酸三叔丁基膦(3.0g,0.01mol,0.03eq)和n,n-二环己基甲胺(80g,0.41mol,1.21eq),搅拌使其混合均匀制得混合液1,混合液1的密度为0.97g/ml,将混合液1放于原料罐1;称取烯丙醇(49g,0.84mol),烯丙醇密度为0.85g/ml,放于原料罐2中;
2、利用本发明的装置图1,按照下述步骤:(1)将原料罐1中混合液1通过计量泵3进入带有t型混合器的管式反应器7,原料罐2中的烯丙醇通过计量泵4进入带有t型混合器的管式反应器7;(2)设定计量泵3和计量泵4控制混合液1和烯丙醇的流速分别为50ml/min和7.0ml/min,设定带有t型混合器的管式反应器7的温度为130℃,反应停留时间5min;(3)反应通道上通过截止阀5和截止阀6来防止物料的倒流;(4)在经过带有t型混合器的管式反应器7混合反应后,粗产物连续出料收集到收集罐8中,粗产物经过hplc分析;(4)粗产物降温至25℃,粗产物中加入乙酸乙酯(700g),过滤除去生成的盐和所述钯催化剂,加入水(200g*3)洗涤有机层,蒸出溶剂,经过蒸馏或重结晶,产物经过gc-ms分析。
3、4-溴-苯甲酸乙酯转化率为99.5%(hplc分析结果);粗产物纯化后,产物纯度为98.2%,收率为89%(gc-ms分析结果)。
- 还没有人留言评论。精彩留言会获得点赞!