具有聚集诱导发光和力致变色特性的化合物及其制备方法和应用与流程
本发明涉及聚集诱导发光材料技术领域,具体涉及一种具有聚集诱导发光和力致变色特性的化合物及其制备方法和应用。
背景技术:
聚集诱导发光(aggregationinduceemission,aie)现象是2001年唐本忠课题组提出来的。众所周知,传统的荧光材料通常在溶液中发光很强,但在固态或者聚集态时发光很弱甚至不发光,这种聚集诱导发光猝灭的现象极大程度的限制了荧光材料的使用。然而,aie材料在溶液中发光很弱,在聚集态或者固态下发光却明显增强。这种aie型材料可以广泛的应用在很多方面,尤其在太阳能电池,oled,生物传感器,多重刺激响应材料等方面得到了很好的应用。
某些材料在受到某些外界刺激时,比如光照、温度、电、压力、酸碱性等时,其光学性质会发生相应的改变,比如发光颜色。这类智能变色材料主要包括光致变色材料、热致变色材料、电致变色材料、气致变色材料、力致变色材料等。其中,力致变色是荧光物质在机械外力的作用下,其发光颜色发生明显变化的现象。在经过诸如加热处理或溶剂熏蒸等过程后,发光颜色又可以恢复到初始状态。由于荧光信号的变化易于检测,力致变色材料在光学记录、传感器、记忆芯片以及光电材料等领域有非常广阔的应用前景,引起了众多研究学者的关注。
上述这类具有力致变色的性能的材料是一类固体材料,而具有聚集诱导发光效应的化合物往往有非常强烈的固态发光,这对于力致变色材料的实际应用是非常有帮助的。到目前为止,已被报道的同时具有聚集诱导发光和力致变色性质的化合物数量是比较有限的。因此,为了提高固体材料的荧光量子效率,以更高效地发挥力致变色材料的性能,研发出一种具有aie性能的力致变色材料是非常有意义的。
技术实现要素:
本发明要解决的技术问题是提供一种具有聚集诱导发光和力致变色特性的化合物,以充分地发挥其力致变色的性能。
为了解决上述技术问题,本发明提供了一种具有聚集诱导发光和力致变色特性的化合物,该化合物含有典型的三苯胺结构,其结构式如式(ⅰ)所示:
式(ⅰ)化合物的制备方法:将双(4-苯甲酰基)苯胺与4-三氟甲基苯甲酰肼在溶剂和催化剂存在的条件下反应,50~60℃下搅拌回流3~6h,得到式(ⅰ)所示的化合物。
本发明还提供了一种具有聚集诱导发光特性的的化合物,该化合物含有典型的三苯胺结构,其结构式如式(ⅱ)所示:
式(ⅱ)化合物的制备方法为:将双(4-苯甲酰基)苯胺与2-三氟甲基苯甲酰肼在溶剂和催化剂存在的条件下反应,50~60℃下搅拌回流3~6h,得到式(ⅱ)所示的化合物。
进一步地,所述溶剂为乙醇或甲醇,所述催化剂为冰醋酸或醋酸酐。
进一步地,所述双(4-苯甲酰基)苯胺与2-三氟甲基苯甲酰肼或4-三氟甲基苯甲酰肼的摩尔比为1:2~1:3。
进一步地,所述搅拌回流的温度为60℃,时间为5h。
进一步地,还包括对反应后的混合液进行过滤、洗涤和重结晶的步骤。优选地,采用乙醇进行洗涤。
进一步地,所述双(4-苯甲酰基)苯胺是经如下的方法制备而成的:
将n,n-二甲基甲酰胺冰水浴至0℃以下,逐滴加入三氯氧磷,搅拌,再加入三苯胺并将混合液加热至90~95℃,搅拌发生反应;反应完成后,将反应液冷却至室温,并倒入冰水中,调至中性,搅拌;经萃取、干燥浓缩后采用色谱柱分离得白色粉末,即为双(4-苯甲酰基)苯胺。
此外,本发明还提供了所述的式(ⅰ)化合物在制备信息存储材料、防伪材料、记忆材料以及力传感材料中的应用。
本发明的有益效果:
1.本发明的化合物的合成方法简单,目标产物在反应过程中直接析出,无需进一步复杂的纯化步骤,合成方式绿色环保,节约原材料。
2.本发明的两种化合物均具有aie性能,实验表明,聚集时的荧光强度分别增强了9倍和8倍,是很好的aie材料。且固态的式(ⅰ)化合物具有力致变色性能,这类刺激响应的固体材料在制备信息存储材料、防伪材料、记忆材料以及力传感材料等方面具有很多潜在的应用。
附图说明
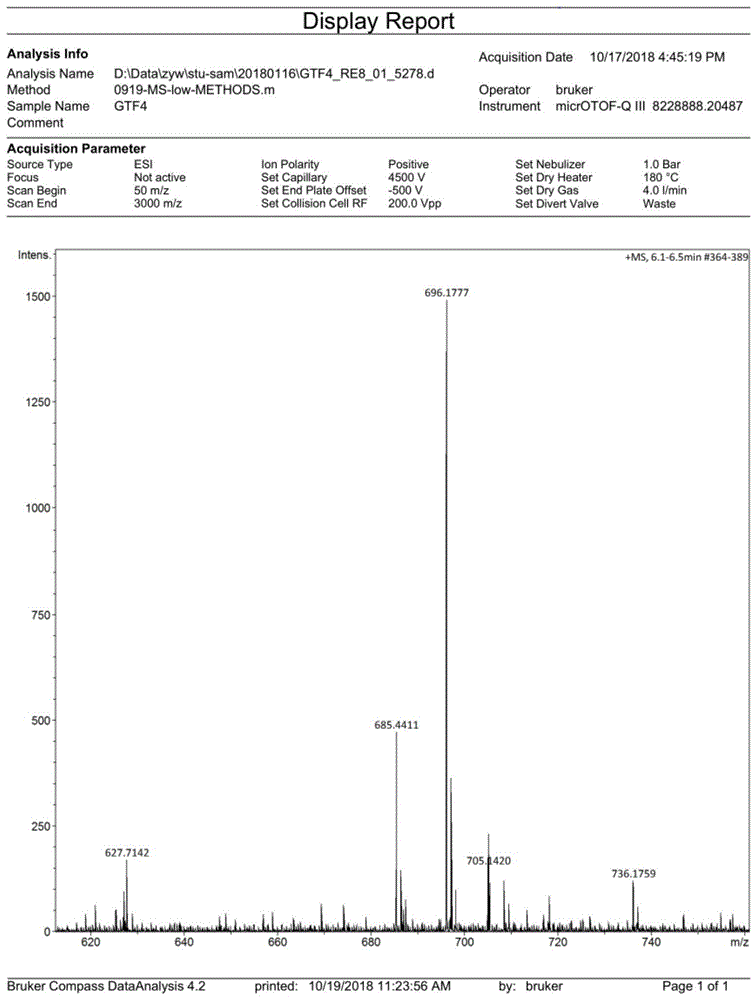
图1是本发明化合物1的质谱图;
图2是本发明化合物2的质谱图;
图3是本发明化合物1的核磁共振氢谱;
图4是本发明化合物2的核磁共振氢谱;
图5是化合物1(a)和化合物2(b)的红外光谱图,化合物1和2的c=n双键分别在1644cm-1和1655cm-1伸缩震动;
图6是化合物1在不同溶剂中的紫外吸收光谱(a)和荧光光谱(b);
图7是化合物2在不同溶剂中的紫外吸收光谱(a)和荧光光谱(b);
图8是化合物1在不同配比的thf/水中的紫外吸收光谱(a)和荧光光谱(b);
图9是化合物2在不同配比的thf/水中的紫外吸收光谱(a)和荧光光谱(b);
图10是化合物1(a)和化合物2(b)的扫描电镜图;
图11是化合物1研磨前(a)和研磨后(b)在自然光下和365nm紫外光下的照片;
图12是化合物1研磨前和研磨后以及溶剂熏后的固体荧光图;
图13是化合物1研磨前、研磨后以及溶剂熏后的粉末衍射测试(wxrd)图;
图14是化合物1和2的的固体荧光图;
图15是化合物1(a)和化合物2(b)的绝对量子产率图;
其中,化合物1为具有式(ⅰ)所示结构的化合物,化合物2为具有式(ⅱ)所示结构的化合物。
具体实施方式
下面结合附图和具体实施例对本发明作进一步说明,以使本领域的技术人员可以更好地理解本发明并能予以实施,但所举实施例不作为对本发明的限定。
本发明各实施例中,乙醇、四氢呋喃、正己烷、甲苯、乙腈、甲醇、二氯甲烷、n,n'-二甲基甲酰胺、氘代dmso均为光谱纯(gr)试剂。三氯氧磷、三苯胺、4-三氟甲基苯甲酰肼、2-三氟甲基苯甲酰肼、氢氧化钠、无水硫酸镁、冰乙酸均为分析纯(ar)。
本发明各实施例中,使用到的检测仪器和检测方法如下:
1、核磁氢谱(1hnmr)是通过(inova400)核磁仪,将待测小分析试样以dmso-d6为溶剂,四甲基硅烷(tms)为内标溶解后进行测试;
2、元素分析是通过yannacochnsocordermt-3元素分析仪来测试;
3、小分子质谱(ms)是通过以乙腈做溶剂,过滤后通过accurate-masstoflc/ms质谱仪测试;
4、熔点是在koflerhb熔点仪上测试的;
5、傅里叶红外变换光谱(ft-tr)测试是在vertex70傅里叶变换红外光谱仪上测的;
6、紫外-可见光谱(uv-vis)测试溶剂效应是配置不同极性的溶剂的化合物溶液(10μm),聚集效应是配置一系列thf和水的溶液(10μm),在cary50上测试;
7、发射光谱的测试,溶剂效应是配置不同极性的溶剂的化合物溶液(10μm),聚集效应是配置一系列thf和水的溶液(50μm),在瞬态/稳态荧光分光光度计(fls920)上测试。
8、扫描电子显微镜是配置聚集时的溶液(50μm),在hitachis4800仪器上测试。
9、相对和绝对荧光量子产率
荧光量子产率是评价荧光材料发光强弱的一个重要的指标之一。单光子荧光量子产率是通过发出的荧光的总量子数与吸收量子数的比值计算的。通常情况下,采用量子计数法直接测量单光子荧光量子产率,然而由于这个实验过程极其复杂,大多数研究者们更愿意采用参比法来测定荧光物质的量子效率。参比法是指在其他条件相同的情况下测定待测化合物与参比物的紫外和荧光光谱,然后通过下式进行计算:
其中u和s分别代表待测物和参比物,φ表示荧光量子产率,i代表荧光积分面积,a为吸光度。在本实验中,以蒽的乙醇溶液(φ=0.27)为参比,以参比物和待测物的紫外光谱图的交点处的吸收峰值作为荧光光谱的激发波长,从而得到参比物和待测物相应的荧光发射谱,将荧光发射光谱的积分面积,带入公式(1)即可以得到各个物质的荧光量子产率。绝对量子效率是由配有积分球的horibafluorolog-3光谱仪测定的。
实施例1
1、合成双(4-苯甲酰基)苯胺
取15.5ml无水dmf冰水浴至0℃以下,逐滴加入15.5ml的三氯氧磷,常温搅拌40min,加入2.0g三苯胺并将混合物加热至95℃,搅拌14小时。反应完成后,将反应物冷却至室温,并倒入冰水中,然后用氢氧化钠调至中性,常温搅拌2小时,用二氯甲烷萃取,无水硫酸镁干燥,浓缩后硅胶柱色谱分离得白色粉末,产率:90%。
1h-nmr(dmso,400mhz)δ(ppm):9.87(s,2h),7.84(d,4h),7.47(t,2h),7.31(s,1h),7.21(d,2h),7.16(d,4h)。
2、合成化合物1
将1mmol双(4-苯甲酰基)苯胺和2mmol4-三氟甲基苯甲酰肼加入到烧瓶中,再加入6ml乙醇和1至2滴冰醋酸。反应在60℃下搅拌回流5小时,后经冷却过滤、乙醇洗涤、重结晶得到黄色固体。
3、合成化合物2
将1mmol双(4-苯甲酰基)苯胺和2mmol4-三氟甲基苯甲酰肼加入到烧瓶中,再加入6ml乙醇和1至2滴冰醋酸。反应在60℃下搅拌回流5小时,后经冷却过滤、乙醇洗涤、重结晶得到黄色固体。
检测与表征
图1~5分别为化合物1和化合物2的质谱图、核磁氢谱图和红外光谱图,下面列出了化合物1和化合物2的元素分析、红外和核磁氢谱的数据。
化合物1:
yield:0.61g(90%).m.p.:312℃.anal.calc.formula:c36h25f6n5o2(%):c:64.19;h:3.74;f:16.92;n:10.40;o:4.75;found:c36h25f6n5o2(%):c:64.11;h:3.80;n:16.94.
ir:1644cm-1(υc=o).
1h-nmr(dmso,400mhz)δ(ppm):12.01(d,j=10.4,1h),11.92(d,j=12.0,1h),8.23(d,j=14.0,1h),8.01(d,j=14.0,1h),7.90(m,10h),7.55(t,j=7.2,1h),7.46(m,2h),7.33(m,3h),7.14(t,j=8.0,2h),7.02(m,3h).hrmscalc.for(m+na+)+:696.1810,found:696.1777.
化合物2:
yield:0.62g(90%).m.p.:271℃.anal.calc.formula:c36h25f6n5o2(%):c:64.19;h:3.74;f:16.92;n:10.40;o:4.75;found:c36h25f6n5o2(%):c:64.25;h:3.70;n:16.90.
ir:1655cm-1(υc=o).
1h-nmr(dmso,400mhz)δ(ppm):11.92(d,j=8.0,1h),11.79(d,j=9.2.0,1h),8.27(d,j=10.8,1h),8.02(d,j=10.4,1h),7.68(m,5h),7.53(t,j=5.6,1h),7.44(m,8h),7.22(m,2h),7.09(d,j=8.4,3h),7.02(m,2h).hrmscalc.for(m+h+)+:674.1946,found:674.1972.
本发明中,将三氟甲基引入腙类结构中,与三苯胺合成具有a-π-d-π-a结构的分子,这种d-a的结构,有利于电荷分离,使分子具有很好的分子内电荷转移(ict)荧光,并且多支的共轭结构,可以使分子发射光谱红移,得到更好的蓝光固体化合物。
图6和图7分别是显示了化合物1和化合物2在甲苯、四氢呋喃、二氯甲烷、乙腈以及甲醇等不同极性溶剂中的紫外吸收和荧光发射光谱图。从吸收光谱图中可以看出,从低极性溶剂甲苯到高极性溶剂甲醇,化合物1和化合物2的最大吸收峰的位置分别处于390nm和383nm附近,极性的变化对最大吸收峰的位置基本不会产生影响,说明化合物1和化合物2基态时电子结构不随极性的改变而改变。从发射光谱上看,化合物1和化合物2随着溶剂极性的增大,光谱红移,化合物1从431nm红移到497nm,化合物2则从420nm红移到488nm,这都表明激发态分子内存在电荷转移荧光。
为了研究两种化合物的聚集诱导发光性能,选择四氢呋喃做良溶剂,水做不良溶剂,测定了两种化合物在不同体积比的四氢呋喃和水溶混液中的紫外吸收和荧光发射光谱图。图8和图9是化合物1和化合物2的聚集效应时的紫外吸收光谱(左)和发射光谱(右)。从吸收光谱上分析,在纯的thf溶剂中,化合物1的最大吸收波长约为389nm,随着含水量的增加,化合物1的最大吸收峰的位置没有明显的变化,当水分数达到90%时,最大吸收峰发生明显红移,吸收光谱出现了明显的拖尾现象,这说明开始形成了聚集,并且聚集尺寸是纳米尺寸。化合物2也发生了类似的变化,纯的thf中在382nm有吸收,当含水量90%时聚集并且有明显的拖尾现象。为了验证在聚集状态下化合物的尺寸是纳米尺寸,对聚集状态下的两种化合物进行了sem测定。图10分别是两种化合物在90%水分数下的sem图像,由此图可以看出化合物1和化合物2的尺寸分别为80-130nm和150-200nm,这说明两种化合物在聚集时的尺寸均是纳米范围内的,与紫外吸收光谱的拖尾现象是相对应的。
研究两种化合物在不同比例的四氢呋喃和水的混合溶液中的荧光行为。化合物1在纯thf溶剂中,相对应的荧光发射光谱,发出较弱的荧光并且最大发射峰451nm,相对其荧光量子产率为4.3%(表1)。在混合溶剂中,当含水量从0增加到70%时,随着溶剂极性增加,荧光强度呈现先降低后增强,含水量达到80%时,荧光强度有开始大幅度增强,表明随着含水量增加发生了聚集,从而使荧光强度逐渐变大。当含水量达到90%时,荧光强度达到最大值约为纯thf溶剂下的9倍(与纯溶剂时相比),最大发射峰红移到493nm,展现了较好的聚集诱导发光增强(aiee)性能,相对量子产率达到了20.9%(表1)。相比化合物1,化合物2聚集过程中,最大发射峰从431nm先红移再蓝移,聚集时在442nm处,聚集时荧光强度增强了8倍,荧光量子产率为16.9%。两种化合物对比,化合物1具有更好的aiee效应。
表1化合物1和化合物2的溶剂效应数据
图10是化合物1和化合物2的扫描电镜图,可以看出聚集态时产生的颗粒是纳米级的(80-130nm,150-200nm)。
经过测试发现,本发明的固体化合物1具有力致变色性能。参见图11,固态化合物1研磨前在自然光下呈浅黄色,经过研磨后,颜色变为黄色。固态化合物1研磨前在365nm紫外光下呈蓝色,经过研磨后,颜色从蓝色变为绿色。
将上述制备好的样品均匀的覆盖在玻璃基底上,测试它们的荧光光谱。如图12所示,化合物1预制样品的最大发射峰在491nm处,经过充分研磨后变527nm,红移36nm。然而,将研磨后样品在有机溶剂二氯甲烷下熏蒸发射峰又恢复至初始状态,这说明化合物1具有良好的力致变色性能。
图13是化合物1研磨前、研磨后以及溶剂熏后的wxrd图,化合物1的初始样品具有比较尖锐、高强度的衍射峰,这说明它拥有高度有序的晶型。然而,经过研磨之后,所得样品的衍射峰强度均大幅度减弱,表明研磨后样品的晶型遭到破坏,是无定形态的。不过,研磨后的样品经过有机溶剂二氯甲烷的熏蒸后,衍射峰又恢复到了初始状态,只是强度有所差异,这说明样品内部发生了分子重排,重新建立了晶体结构,这种转变是晶态到非晶态的转变。
图14是化合物1和化合物2的固体荧光图。相比化合物1,化合物2研磨后没有表现出力致变色性能,化合物1固体的在最大发射峰在491nm,化合物2的最大发射峰在475nm,相比较而言,化合物1发射峰稍微红移。
另外,图15是两种化合物的固体荧光量子产率图,化合物1的绝对荧光量子产率为8.08%,化合物2的荧光量子产率为3.68%。综合比较,化合物1的固体发光性能比化合物2好。
以上测试表明,研磨使粉末从一种热稳定状态转变为一种亚稳定状态,而有机溶剂熏蒸则使热稳定状态得到恢复。这类刺激响应的固体材料有很多潜在的应用,使其在光学领域的应用得到延伸。
以上所述实施例仅是为充分说明本发明而所举的较佳的实施例,本发明的保护范围不限于此。本技术领域的技术人员在本发明基础上所作的等同替代或变换,均在本发明的保护范围之内。本发明的保护范围以权利要求书为准。
- 还没有人留言评论。精彩留言会获得点赞!