一种盐酸帕唑帕尼原料药三聚体杂质的合成方法与流程
本发明属于药物化学领域,特别涉及一种盐酸帕唑帕尼原料药三聚体杂质的合成方法。
背景技术:
盐酸帕唑帕尼,化学名为5-({4-〔(2,3-二甲基-2h-吲唑-6-基)(甲基)氨基〕-嘧啶-2-基}氨基)-2-甲基苯磺酰胺盐酸盐,分子式为c21h23n2o2s·hcl,分子量为473.98,其结构式如下:
盐酸帕唑帕尼是由英国葛兰素史克研发的一种血管内皮生长因子受体(vegfr)的酪氨酸激酶抑制剂,通过选择性抑制vegfr-1,vegfr-2,vegfr-3与atp竞争性结合胞外的配体结合位点,阻断内酪氨酸的自身硫酸化,抑制vegfr激活,从而加速细胞凋亡,抑制血管生成,抑制肿瘤浸润和转移,研究发现盐酸帕唑帕尼对治疗晚期肾细胞癌,软组织肉瘤,晚期卵巢癌等疾病有良好的疗效。
在药物开发过程中,药品的安全性是评价药品质量好坏的重要依据;由于杂质可能对人体有潜在的毒性,所以杂质研究是药物研究开发的重要组成部分。近年来,频频爆发的药品质量安全事故,越来越引起国家对药品质量的重视。药品杂质本身由于含量很低,提取和合成难度较大,分离和结构鉴定存在较大困难。另外,杂质用量较小,不易规模化生产,产生的效益较小,因此需要投入大量人力物力进行研究开发。
盐酸帕唑帕尼三聚体杂质作为盐酸帕唑帕尼原料药中的一种重要有关物质,目前没有检索到该杂质的合成方法,为了保证后续物质的对照,需要研究合理的方法制得该杂质的标准品,同时该杂质成功的合成,也能对提高盐酸帕唑帕尼原料药的质量起到很大的推动作用。
技术实现要素:
针对上述问题,本发明提供一种盐酸帕唑帕尼原料药三聚体杂质的合成方法,所述三聚体杂质具有如下式(v)的结构:
所述式(v)三聚体杂质的合成方法包括:
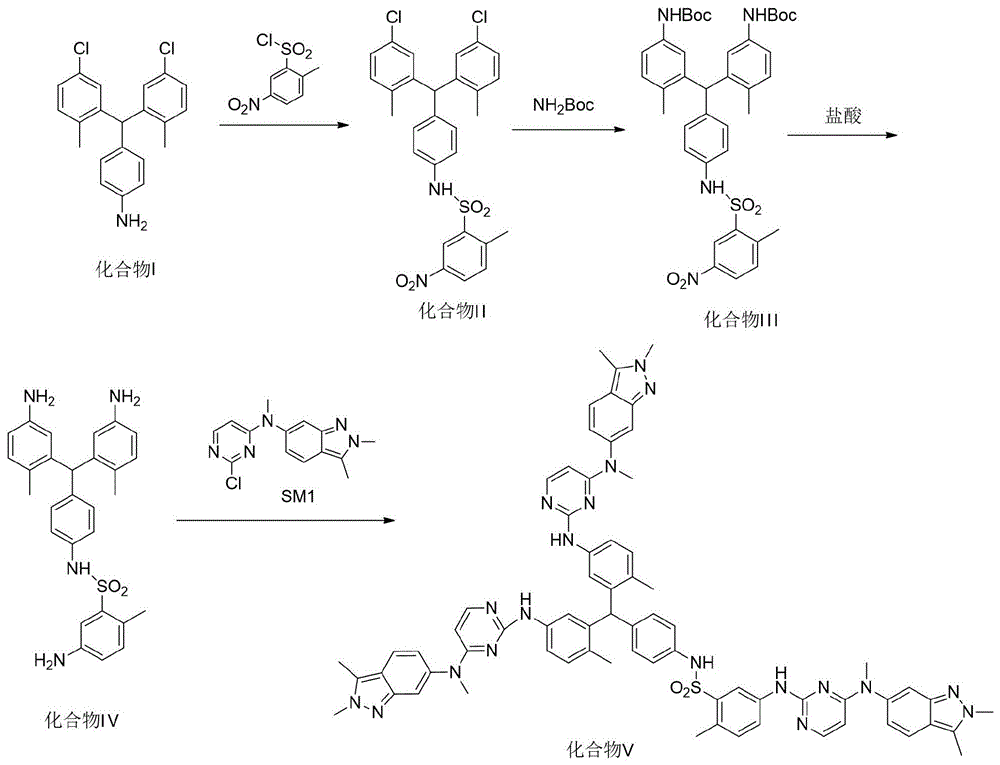
步骤一:以化合物i为原料,使之与2-甲基-5-硝基苯磺酰氯缩合反应得化合物ii;
步骤二:取化合物ii,使之与氨基甲酸叔丁酯反应生成化合物iii;
步骤三,取化合物iii,使之在盐酸条件下反应脱去保护基,得到化合物iv;
步骤四,取化合物iv,使之与n-(2-氯嘧啶-4-基)-n-甲基-2,3-二甲基-2h-吲唑-6-胺进行缩合反应,得到三聚体杂质化合物v。
进一步地,所述步骤一中缩合反应的溶剂包括丙酮、四氢呋喃、二氧六环的一种或多种,所述缩合反应的缚酸剂包括三乙胺、吡啶、氢氧化钠、氢氧化钾和碳酸钾中的一种或多种。
进一步地,所述步骤二中反应的溶剂包括丙酮、二氯甲烷、氯仿、四氢呋喃中的一种或多种,所述反应的催化剂为三苯基膦钯。
进一步地,所述步骤三中脱去保护基是在质子性溶剂中,所述质子性溶剂包括甲醇、乙醇、异丙醇、正丁醇中的一种或多种;所述反应温度为0~50℃。
进一步地,所述步骤四中缩合反应的反应溶剂包括n,n-二甲基甲酰氨、n,n-二甲基乙酰氨、四氢呋喃、二氧六环、丙酮中的一种或多种以及它们中的一种或多种与甲醇、乙醇、异丙醇、正丁醇、水中的一种或多种混合;所述反应的催化剂包括盐酸、对甲苯磺酸、磷酸、硫酸、甲磺酸中的一种或多种。
本发明的一种盐酸帕唑帕尼原料药三聚体杂质的合成方法,合成的收率高,制备得到的杂质可为盐酸帕唑帕尼的质量分析提供对照品,从而提升盐酸帕唑帕尼的质量标准。
本发明的其它特征和优点将在随后的说明书中阐述,并且,部分地从说明书中变得显而易见,或者通过实施本发明而了解。本发明的目的和其他优点可通过在说明书、权利要求书以及附图中所指出的结构来实现和获得。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作一简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
图1示出了根据本发明实施例的合成流程图。
具体实施方式
为使本发明实施例的目的、技术方案和优点更加清楚,下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地说明,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
本发明提供一种盐酸帕唑帕尼原料药三聚体杂质的合成方法,所述盐酸帕唑帕尼原料药三聚体杂质的结构式如下式(v):
图1示出了根据本发明实施例的合成流程图。如图1所示,所述盐酸帕唑帕尼原料药三聚体杂质式(v)的合成方法包括:
步骤一:取化合物i,使之与2-甲基-5-硝基苯磺酰氯缩合反应得化合物ii;
具体的,所述缩合反应的溶剂包括丙酮、四氢呋喃、二氧六环的一种或多种,优选的,所述缩合反应的溶剂为四氢呋喃,所述缩合反应的缚酸剂包括三乙胺、吡啶、氢氧化钠、氢氧化钾和碳酸钾中的一种或多种,优选的,所述缚酸剂为吡啶。
步骤二:取化合物ii,使之与氨基甲酸叔丁酯反应生成化合物iii;
具体的,所述反应的溶剂包括丙酮、二氯甲烷、氯仿、四氢呋喃中的一种或多种,所述反应的催化剂为三苯基膦钯。
步骤三,取化合物iii,使之在盐酸条件下反应脱去保护基,得到化合物iv;
具体的,所述脱去保护基是在质子性溶剂中,所述质子性溶剂包括甲醇、乙醇、异丙醇、正丁醇中的一种或多种;所述反应温度为0~50℃。
步骤四,取化合物iv,使之与sm1(n-(2-氯嘧啶-4-基)-n-甲基-2,3-二甲基-2h-吲唑-6-胺)进行缩合反应,得到三聚体杂质化合物v。
具体的,所述缩合反应的反应溶剂为n,n-二甲基甲酰氨、n,n-二甲基乙酰氨、四氢呋喃、二氧六环、丙酮中的一种或多种和它们中的一种或多种与甲醇、乙醇、异丙醇、正丁醇、水中的一种或多种的混合,这二者形成混合溶液为反应溶剂,在室温和回流条件下,以盐酸、对甲苯磺酸、磷酸、硫酸、甲磺酸中的一种或多种为催化剂的条件下,与sm1(n-(2-氯嘧啶-4-基)-n-甲基-2,3-二甲基-2h-吲唑-6-胺)反应,合成化合物v。
其中,sm1(n-(2-氯嘧啶-4-基)-n-甲基-2,3-二甲基-2h-吲唑-6-胺)的结构式为:
实施案例一,化合物ii的合成方法为:
取100ml两口反应瓶,分别加入4.05g的化合物i、40ml的四氢呋喃和5ml的吡啶,并加入2-甲基-5-硝基苯磺酰氯(2.95g,12.51mmol,1.1eq),而后在室温下搅拌,搅拌充分后放置过夜;而后在反应液中加入乙酸乙酯和水,萃取分液,分液后取得的有机相用饱和食盐水洗涤,无水硫酸钠干燥,过滤,旋蒸至干;得到的粗品过硅胶柱纯化,得到3.85g黄色固体;收率:61%。
上述反应式为:
实施案例二,化合物iii的合成方法为:
取100ml两口反应瓶,氮气保护下,分别加入3.85g的化合物ii、50ml的四氢呋喃、0.1g的三苯基膦钯、2.50g的碳酸钾和适量的氨基甲酸叔丁酯,加热升温至70℃反应,反应结束后,旋蒸除去溶剂,然后再加入乙酸乙酯和水,萃取分液,分液后取得的有机相用饱和食盐水洗涤,无水硫酸钠干燥,过滤,旋蒸至干,得到的粗品过硅胶柱纯化,得到5.07g黄色色固体;收率:98%。
上述反应式为:
实施案例三,化合物iv的合成方法为:
取100ml两口反应瓶,在反应瓶中分别加入5.07g的化合物iii、50ml的二氧六环、5ml的浓盐酸和锌粉(2.00g,30.59mmol,4.3eq),并在室温下搅拌反应,反应结束后,旋蒸除去溶剂,向残余物中加入乙酸乙酯和水,萃取分液,分液后取得的有机相用饱和碳酸氢钠洗涤,饱和食盐水洗涤,无水硫酸钠干燥,过滤,浓缩,得到2.90g淡黄黄色固体;收率:84%。
上述反应式为:
实施案例四,化合物v的合成方法为:
取100ml两口反应瓶,分别加入0.50g的化合物iv、50ml的无水甲醇和sm1(1.77g,6.16mmol,6eq),加热升温至回流,反应结束后,旋蒸除去溶剂,将得到的粗品过硅胶柱纯化,得到1.05g淡黄色固体;收率:83%。
上述反应式为:
尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的精神和范围。
- 还没有人留言评论。精彩留言会获得点赞!