双交联胶原水凝胶材料、其制备方法与应用与流程
本发明涉及一种水凝胶材料,具体涉及一种双交联胶原水凝胶材料、其制备方法与应用,生物材料技术领域。
背景技术:
3d打印技术,又名快速成型、固体自由制造、增材制造等。随着3d打印技术的进一步发展和成熟,3d打印与材料科学,生物科学等多学科相结合,在组织工程的研究当中也进行了广泛的应用,如制造供临床使用的支架或者夹板等医疗器械。
在3d生物打印的过程中,生物打印材料作为构建三维支架的基础,选择合适的材料进行3d打印,是其中的关键一步。总体而言,选用的生物材料必须具备三个基本特征:
(1)生物相容性。打印材料必须是对细胞友好,在体内不会发生免疫反应,最好是有利于细胞的黏附增殖。
(2)可打印性。打印材料可以在打印机中被打印成型,材料的黏度,流变学特性等适于被打印机打印成型。
(3)适宜的力学强度。选用的材料必须具备一定的力学强度来构建三维支架,保证在打印的过程中不坍塌变形,能够达到特定的组织的力学强度要求。
作为细胞外基质的一部分,胶原蛋白对周围细胞的结构和生物功能的起支撑作用,并广泛存在于软硬结缔组织中,其可以用于3d打印制备组织工程支架。但胶原应用于组织工程也存在着一些亟待解决的问题,例如力学强度低、稳定性差、易降解、不易打印等。因此,目前胶原参与的3d打印过程一般为复合型材料的打印,在打印过程中需要和其他材料发生相互作用或者添加一些有毒性的交联剂来进行交联成型。直接单独利用胶原进行3d打印,制备特定的机械强度适宜的组织工程三维支架的相关报道较少。
技术实现要素:
本发明的主要目的在于提供一种双交联胶原水凝胶、其制备方法与应用,从而克服现有技术中的不足。
为实现前述发明目的,本发明采用的技术方案包括:
本发明实施例提供了一种双交联胶原水凝胶的制备方法,其包括:在有tg酶存在的条件下,使乌头酸修饰的α-β不饱和羰基化胶原与巯基化胶原进行michael加成反应和酶催化反应,从而获得双交联胶原水凝胶。
在一些实施方案中,所述的制备方法具体包括:先以还原剂对所述巯基化胶原进行处理,之后与乌头酸修饰的α-β不饱和羰基化胶原在有tg酶存在的条件下进行michael加成反应和tg酶催化反应。
其中,所述乌头酸修饰的α-β不饱和羰基化胶原的通式为:
所述巯基化胶原的通式为:
以上各式中,r代表胶原去除氨基和羧基后的残基部分,r1为氢原子、烷基或亚烷基,r2为氢原子或羧基或羟基衍生物,r3为亚烷基或取代亚烷基。
进一步的,所述乌头酸修饰的α-β不饱和羰基化胶原的制备方法包括:将胶原的酸溶液和乌头酸溶液混合后进行反应,混合反应的温度在37℃以下,之后进行透析、冷冻干燥,其中胶原链上游离氨基与乌头酸的摩尔比为1:1~10。
进一步的,所述巯基化胶原的制备方法包括:使胶原的酸溶液与巯基化合物溶液在羧基活化剂的作用下进行混合反应,混合反应的温度在37℃以下,之后进行透析、冷冻干燥,其中所述巯基化合物与所述胶原上氨基或羧基的摩尔比为1~10:1。
本发明实施例还提供了一种双交联胶原水凝胶,其由前述的任一种方法制备。
本发明实施例还提供了所述双交联胶原水凝胶的用途,例如在制作生物医疗材料、组织工程材料或3d打印材料中的应用。
较之现有技术,本发明的优点包括:
(1)本发明实施例提供的双交联胶原水凝胶的制备方法中,通过将乌头酸α-β不饱和羰基化的胶原与巯基化的胶原作为原材料,利用michael加成反应和tg酶催化反应,在4℃左右的打印条件下可实现原材料由液态到固态的快速转变,反应迅速,条件温和,操作简单,不会出现明显的副反应产物,大大提高了胶原材料的可打印性,通过调节二者的浓度、小分子的接枝率和tg酶的加入量等条件可实现固化后水凝胶弹性模量的调节,可扩大胶原材料的应用范围。
(2)本发明实施例提供的双交联胶原水凝胶的多项性能均得到了显著提升,,其固化速度快,生物相容性好、机械强度可调、在培养基中的稳定性好,生物降解速度可调,在生物医学及组织工程等领域具有重要用途。
附图说明
为了更清楚地说明本发明实施方式的技术方案,下面将对实施方式中所需要使用的附图作简单地介绍,应当理解,以下附图仅示出了本发明的某些实施例,因此不应被看作是对范围的限定,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他相关的附图。
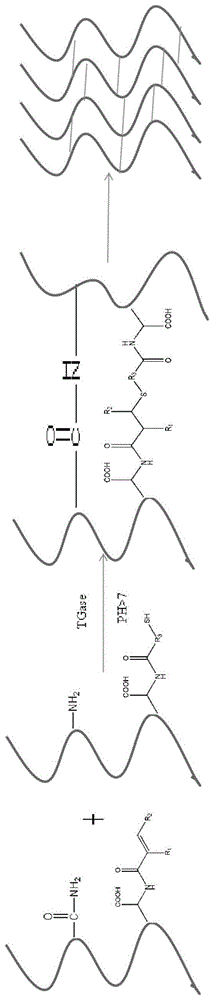
图1为本发明一典型实施方案中一种双交联胶原水凝胶的合成工艺原理图。
图2为本发明实施例1中胶原与巯基化胶原的傅里叶红外图谱(ftir)对比分析图。
图3为本发明实施例1中胶原与乌头酸修饰胶原的傅里叶红外图谱(ftir)对比分析图。
图4为本发明实施例1中胶原与制备的胶原双交联水凝胶的傅里叶红外图谱(ftir)对比分析图。
图5为本发明实施例1中的巯基化胶原的扫描电子显微镜(sem)表面微观结构图。
图6为本发明实施例1中的乌头酸α-β不饱和羰基化胶原产物的扫描电子显微镜(sem)表面微观结构图。
图7为本发明实施例1中固化后水凝胶的扫描电子显微镜(sem)表面微观结构图。
图8为本发明实施例1中固化后水凝胶的表面孔径扫描电子显微镜(sem)图。
图9为本发明实施例1中巯基化胶原的cd图。
图10为本发明实施例1中乌头酸α-β不饱和羰基化胶原cd图。
图11为本发明实施例1中固化后水凝胶的力学性能测试图。
具体实施方式
鉴于现有技术的不足,本案发明人经长期研究和大量实践,得以提出本发明的技术方案。概括地讲,本发明是通过对胶原材料进行化学改性,使其在打印过程中能够进行化学交联,进而实现基于胶原的3d打印生物材料的固化成型。如下将对本发明的技术方案进行更为具体的解释说明。
本发明实施例的一个方面提供了一种双交联胶原水凝胶,其成胶机理可以参阅附图1。概括的讲,所述双交联胶原水凝胶可以由乌头酸修饰的α-β不饱和羰基化胶原与巯基化胶原通过michael加成反应和酶催化反应形成。
在图1中,所示的r1可以选自氢原子、取代或未取代的烷基或亚烷基,r2可以选自氢原子、羧基或者羧基衍生物。
进一步的,r1所含的碳原子数均可为1~20个。即r1可以为c1、c2、c3、c4、c5、c6、c7、c8、c9、c10、c11、c12、c13、c14、c15、c16、c17、c18、c19、c20的亚烷基或取代亚烷基。
本发明实施例的另一个方面还提供了一种双交联胶原水凝胶的制备方法,其包括:在有tg酶存在的条件下,使乌头酸修饰的α-β不饱和羰基化胶原(下文亦简称为“乌头酸修饰胶原”)与巯基化胶原进行michael加成反应和酶催化反应,从而获得双交联胶原水凝胶。
在一些实施方案中,所述的制备方法可以包括:将混合了tg酶的乌头酸修饰的α-β不饱和羰基化胶原与巯基化胶原进行michael加成反应和酶催化反应。
在一些实施方案中,所述的制备方法可以包括:先以还原剂对所述巯基化胶原进行处理,之后与乌头酸修饰的α-β不饱和羰基化胶原在有tg酶存在的条件下进行michael加成反应和tg酶催化反应。
进一步的,所述还原剂包括金属zn、二硫苏糖醇或对苯二酚中的一种或多种,且不限于此。
在一些实施方案中,所述的制备方法可以包括:将tg酶、乌头酸修饰的α-β不饱和羰基化胶原的溶液与巯基化胶原的溶液混合进行所述的michael加成反应和tg酶催化反应。进一步的,所述乌头酸修饰的α-β不饱和羰基化胶原的溶液的浓度为10~100mg/ml,ph值小于7。
进一步的,所述巯基化胶原溶液的浓度为10~100mg/ml,ph值大于7。
更进一步的,在本发明的一些实施方式中,乌头酸修饰胶原溶液是将乌头酸修饰胶原溶解在酸溶液而配成的浓度为10~100mg/ml的乌头酸修饰胶原酸溶液。巯基化胶原溶液是将巯基化胶原溶解在碱溶液中而配成的浓度为10~100mg/ml的巯基化胶原碱溶液。通过上述浓度以及其溶剂的设置,使得乌头酸修饰胶原以及巯基化胶原能够很好的进行溶解,并且能够更加有利于二者之间的michael加成反应和酶催化交联反应的进行。在一些实施方案中,所述的制备方法具体包括:将所述乌头酸修饰的α-β不饱和羰基化胶原的溶液与所述巯基化胶原的溶液、tg酶直接混合而进行所述的michael加成反应和tg酶催化反应。
在一些实施方案中,所述的制备方法具体包括:使所述乌头酸修饰的α-β不饱和羰基化胶原的溶液、巯基化胶原的溶液与tg酶的混合溶液在被打印设备或挤出成型设备挤出的过程中相互接触而进行所述的michael加成反应和tg酶催化反应。
在一些实施方案中,所述乌头酸修饰的α-β不饱和羰基化胶原的制备方法包括:将胶原的酸溶液和乌头酸溶液混合后进行反应,混合反应的温度在37℃以下,之后进行透析、冷冻干燥,其中胶原链上游离氨基与乌头酸的摩尔比为1:1~10。
在一些实施方案中,所述巯基化胶原的制备方法包括:使胶原的酸溶液与巯基化合物溶液在羧基活化剂的作用下进行混合反应,混合反应的温度在37℃以下,之后进行透析、冷冻干燥,其中所述巯基化合物与所述胶原上氨基或羧基的摩尔比为1~10:1。
其中,所述透析的截留分子量为≤7000da。
其中,所述冷冻干燥的温度在≤-80℃。
在一些实施方案中,在所述的制备方法中,所述混合反应的温度为10~30℃,优选为15~20℃。
在一些实施方案中,所述胶原的酸溶液是通过将胶原溶解于浓度为0.01~30wt%的酸溶液中形成。
在一些实施方案中,所述巯基化合物溶液是通过将巯基化合物溶解于双蒸水、碱溶液或酸溶液中形成。
在一些实施方案中,所述羧基活化剂包括质量比为1~10:1的edc和nhs。
在一些实施方案中,所述巯基化合物为同时含有羧基和巯基或同时含有氨基和巯基的化合物。上述巯基化合物可以通过经水解反应、氨解反应等直接生成,或者可通过含二硫键的化合物经还原后得到。例如,同时具有羧基和巯基的化合物可以为:二巯基丁二酸、巯基丁二酸、巯基丙酸、硫代乙醇酸、2-巯基-3-吡啶甲酸等。同时具有氨基和巯基的化合物可以为:半胱氨酸甲酯、半胱氨酸乙酯、4-氨基-3-巯基苯甲酸等,但不限于此。在一些实施方案中,所述巯基化胶原的制备方法还包括:先利用羧基活化剂对巯基化合物或胶原中的羧基进行活化,再将巯基化合物溶液与胶原的酸溶液进行混合反应。
在本发明的前述实施例中,用以进行巯基化的胶原可以选自胶原蛋白、碱性明胶蛋白、酸性明胶蛋白、基因重组胶原蛋白、碱性基因重组明胶蛋白、酸性基因重组明胶蛋白、核心蛋白、多糖层粘连蛋白以及纤维结合蛋白中的一种。这些分子链上存在游离的氨基和羧基,从而可以通过与羧基或氨基进行反应,达到对胶原分子链进行修饰改性的目的。进一步的,所述乌头酸修饰的α-β不饱和羰基化胶原的通式为:
进一步的,所述巯基化胶原的通式为:
其中,r代表胶原去除氨基和羧基后的残基部分,r1为氢原子、烷基或亚烷基,r2为氢原子或羧基或羟基衍生物,r3为亚烷基或取代亚烷基。
进一步的,前述取代或未取代的烷基或亚烷基所含的碳原子数可为1~20个,即可以为c1、c2、c3、c4、c5、c6、c7、c8、c9、c10、c11、c12、c13、c14、c15、c16、c17、c18、c19、c20的烷基或亚烷基。
进一步的,前述取代的烷基或亚烷基可以为至少一个氢原子被烷基、羧基、氨基、烷氧基、芳香基、酯基和卤代烷基中至少一种基团取代的烷基或亚烷基。即,可以是烷基或亚烷基中有一个氢原子被烷基、羧基、氨基、烷氧基、芳香基、酯基和卤代烷基中的一种基团取代;也可以是烷基或亚烷基中有两个氢原子被烷基、羧基、氨基、烷氧基、芳香基、酯基和卤代烷基中的两种基团取代;也可以是烷基或亚烷基中有两个以上的氢原子被烷基、羧基、氨基、烷氧基、芳香基、酯基和卤代烷基中的两种以上的基团取代;也可以是烷基或亚烷基中多个氢原子被烷基、羧基、氨基、烷氧基、芳香基、酯基和卤代烷基中的多个同一种基团取代或被其中多个不同基团的组合对应进行取代。
在本发明的前述实施例中,采用的胶原具有良好的生物相容性、生物降解性、杀菌性等,是一种安全的天然高分子聚合物。胶原分子上的游离氨基具有较高的化学活性,易于被羧基等活性较高的官能团修饰,利用乌头酸对胶原进行修饰,可得到水溶性胶原,同时在胶原分子链上引入了α-β不饱和羰基结构,可以被亲核试剂进攻发生加成反应,成功实现聚合物的交联,达到快速固化的效果。
更具体地讲,胶原是一类重要的蛋白质,通常由三条肽链组成,这些肽链被称为α-链。胶原是细胞外基质中的一种结构蛋白,按照其结构功能的划分,可分为i、ii、iii、iv等型胶原。胶原水凝胶已经成功实现了细胞在三维基板上的培养,在组织工程中也被认为是一种非常有应用前景的支架材料。虽然胶原本身可以形成水凝胶,但由于其独立形成的水凝胶机械强度较弱,弹性模量较小,且交联方式不稳定,分子链间作用力在培养基中容易被破坏而溶解,限制了其应用。胶原分子链上存在游离氨基基团和羧基基团,用带巯基的化合物去修饰氨基或羧基,可将胶原巯基化,修饰后的胶原具备巯基的化学性质,在碱性条件下巯基质子离去可产生硫负离子,作为亲核试剂进攻用乌头酸α-β不饱和羰基化的胶原,以c-s键的形式将巯基化胶原和乌头酸α-β不饱和羰基化胶原快速接枝在一起,生成网状水凝胶。
在本发明的一些较为具体的实施方式中,所述水凝胶的制备方法包括如下步骤进:
(1)制备乌头酸修饰胶原(col-taa)。
乌头酸修饰胶原的化学反应式为:
根据一些实施方式,可以将胶原溶解在酸溶液中,将乌头酸溶解于极性溶剂中,胶原链上游离氨基与乌头酸的摩尔比优选为1:1~3,将溶解的乌头酸在磁力搅拌器上缓慢加入胶原酸溶液中,室温下充分混合均匀,优选地,反应温度为37℃以下,更优选为10~30℃,进一步优选为15~20℃,反应时间为4~12h。反应结束后经过透析2~4天,冷冻干燥2~4天,得到修饰后胶原产物。
根据一些实施方式,用以溶解胶原的酸溶液可以是有机酸溶液,优选乙酸溶液,更优选地,乙酸溶液的质量分数为0.01~30%。溶解乌头酸的极性溶剂包括丙酮、丁酮、水、dmso、dmf等,优选丙酮,且丙酮的用量为保证将乌头酸完全溶解即可。通过将胶原链上游离氨基与乌头酸的摩尔比设置为1:1~3,使得乌头酸能够更好的与胶原链上游离氨基进行反应,从而对胶原修饰得到乌头酸修饰胶原。同时,将溶解的乌头酸在磁力搅拌器上搅拌,并缓慢加入胶原酸溶液中,能够使得二者能够更加充分地进行接触,进而达到更好的反应效果。在反应结束后对反应液进行透析和冷冻干燥操作,可以将乌头酸修饰胶原中残留的小分子杂质更好的去除,以得到纯度更高的乌头酸修饰胶原,进而促进后续michael加成反应的更好进行。
(2)制备巯基化胶原(col-sh)。
巯基化胶原(col-sh)可以由以下反应式生成:
或
根据一些实施方式,巯基化胶原(col-sh)的制备方法是将含有胶原的溶液与含有巯基化合物的溶液在羧基活化剂的作用下进行反应。
根据一些实施方式,当采用含有羧基的巯基化合物作为原料时,巯基化胶原(col-sh)可以通过以下方法制备:
a.制备含有胶原的溶液:将胶原溶解于质量分数为0.01~30%的酸溶液中。
b.制备含巯基化合物的溶液:将巯基化合物,根据其溶解性,溶解于双蒸水或碱溶液或酸溶液中。
c.将羧基活化剂加入到上述含巯基化合物的溶液中,混合均匀后,调节其ph值至4.5~6.5,继续混合搅拌。
d.将活化后的含有巯基化合物的溶液与含有胶原的溶液进行混合,巯基化合物与胶原上氨基的摩尔比为1~10,充分搅拌均匀,优选转移至圆底烧瓶中,放置在温度为37℃以下进行恒温反应。
e.将反应后得到的反应液进行透析2~5天,再将得到的透析产物进行冷冻干燥2~5天后,得到巯基化胶原。通过透析的手段可以除去残留的小分子杂质,提高所得的胶原巯基化衍生物的纯度。
需要说明的是,上述过程中也可以先进行步骤b和c,再进行步骤a,其不会影响最终产物的生成。
根据一些实施方式,巯基化胶原(col-sh)制备过程中的酸溶液可以是有机酸溶液,优选乙酸溶液。即优选将胶原溶解于质量分数为0.01~30%的乙酸溶液中。
根据一些实施方式,碱溶液可以为强碱溶液,例如氢氧化钠溶液、氢氧化钾溶液、氢氧化钙溶液等,也可以为弱碱溶液,例如氨水溶液、碳酸钠溶液、碳酸氢钠溶液等,优选氢氧化钠溶液。双蒸水可以使得溶解后的溶液杂质含量少,纯度更高,后续反应效果越好。溶解巯基化合物的酸溶液可以是有机酸溶液,优选乙酸溶液。
根据一些实施方式,羧基活化剂包括edc和nhs,edc即1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐,edc是可溶于水的碳二亚胺,在酰胺合成中用作羧基的活化试剂,也用于活化磷酸酯基团、蛋白质与核酸的交联和免疫偶联物的制取,常和n-羟基琥珀酰亚胺(nhs)或n-羟基硫代琥珀酰亚胺连用,以提高偶联效率。nhs即n-羟基琥珀酰亚胺,活化羧基以利于酰胺键的形成。用于合成氨基酸保护剂、半合成卡那霉素及医药中间体。本发明实施例中的羧基活化剂可按照edc和nhs的质量比为1~10:1的比例进行配制,这种比例有利于二者达到更好的偶联效率,使得对巯基化合物的羧基活化效果更好。
优选地,可以将edc和nhs在磁力搅拌器作用下加入到上述含巯基化合物的溶液中,以达到更好的混合效果。
根据一些实施方式,混合羧基活化剂后,用碱或酸溶液调节加有羧基活化剂的巯基化合物溶液的ph值,使得其ph值为4.5~6.5。优选地,用1m氢氧化钠或1m盐酸溶液进行调节ph值。edc和nhs在ph值为4.5~6.5的条件下能够达到对羧基最好的活化效果。
根据一些实施方式,调节ph值后混合搅拌的时间为10~150min,使得羧基活化剂能够对巯基化合物的羧基进行充分的活化,以更好地对胶原进行修饰。
其中,反应温度优选10~30℃,进一步优选15~20℃,反应时间可以为1~8h,优选2~6h,更优选地4~5h。
在本发明其它较佳实施方式中,当以含有氨基的巯基化合物作为原料时,胶原巯基化衍生物还可以通过以下方法制备:
a.制备含有胶原的溶液:将胶原溶解于质量分数为0.01~30%的酸溶液中。
根据一些实施方式,酸溶液可以是有机酸溶液,优选乙酸溶液。即优选将所述胶原溶解于质量分数为0.01~30%的乙酸溶液中。
b.制备含巯基化合物的溶液:将巯基化合物,根据其溶解性,溶解于双蒸水或碱溶液或酸溶液中。
根据一些实施方式,碱溶液可以为强碱溶液,例如氢氧化钠溶液、氢氧化钾溶液、氢氧化钙溶液等,也可以为弱碱溶液,例如氨水溶液、碳酸钠溶液、碳酸氢钠溶液等,优选氢氧化钠溶液。双蒸水可以使得溶解后的溶液纯度更高,后续反应效果越好。溶解巯基化合物的酸溶液可以是有机酸溶液,优选乙酸溶液。
c.将羧基活化剂加入到上述含胶原的溶液中,混合均匀后,调节其ph值至4.5-6.5,继续混合搅拌。
根据一些实施方式,羧基活化剂包括edc和nhs。本发明实施方式中的羧基活化剂可按照edc和nhs的质量比为10:1~1:1的比例进行配制,这种比例有利于二者达到更好的偶联效率,使得对胶原的羧基活化效果更好。
根据一些实施方式,可以取edc和nhs在磁力搅拌器作用下加入到上述含胶原的溶液中,以达到更好的混合效果。
根据一些实施方式,混合羧基活化剂后,用碱或酸溶液调节加有羧基活化剂的胶原溶液的ph值,使得其ph值为4.5-6.5。优选地,用1m氢氧化钠或1m盐酸溶液进行调节ph值。edc和nhs在ph值为4.5-6.5的条件下能够达到对羧基最好的活化效果。
根据一些实施方式,调节ph值后混合搅拌的时间为10~150min,使得羧基活化剂能够对胶原的羧基进行充分的活化,以更好地对胶原进行修饰。
d.将活化后的含有巯基化合物的溶液与含有胶原的溶液进行混合,巯基化合物与胶原上羧基的摩尔比为1~10:1,充分搅拌均匀,搅拌时间优选10~150min,且优选将混合液转移至圆底烧瓶中,放置在温度为37℃以下进行恒温反应。
根据一些实施方式,反应温度优选10~30℃,进一步优选15~20℃,反应时间可以为1~8h,优选2~6h,更优选地4~5h,其中,反应温度不限于37℃以下。
e.将反应后得到的反应液进行透析2~5天,再将得到的透析产物进行冷冻干燥2~5天后,得到胶原巯基化衍生物。通过透析的手段可以除去残留的小分子杂质,提高所得的胶原巯基化衍生物的纯度。
需要说明的是,上述过程中也可以先进行步骤b,再进行步骤a,其不会影响最终产物的生成。
(3)将乌头酸修饰胶原溶液与巯基化胶原溶液进行michael加成反应和酶催化交联反应。
根据一些实施方式,进行michael加成反应和酶催化交联反应可以是将乌头酸修饰胶原溶液与巯基化胶原溶液直接混合tg酶成型或利用3d打印机进样,并挤出接触成型。通过3d打印机来混合乌头酸修饰胶原溶液与巯基化胶原溶液使得二者可以按照一定比例进行充分混合,从而更加有利于反应的进行以及对生成的水凝胶的性能的调节。
具体地,首先,将步骤(1)得到的乌头酸修饰胶原溶解在质量分数为0.1~30%的酸溶液中,配成浓度10~50mg/ml的溶液;其中,酸溶液可以有机酸溶液,优选乙酸溶液。其次,将步骤(2)得到的巯基化胶原溶解在碱溶液中配成10~50mg/ml的溶液,并将溶液中的巯基化胶原经还原剂处理。
其中,碱溶液可以为强碱溶液,例如氢氧化钠溶液、氢氧化钾溶液、氢氧化钙溶液等,也可以为弱碱溶液,例如氨水溶液、碳酸钠溶液、碳酸氢钠溶液等,优选氢氧化钠溶液。还原剂为金属zn、二硫苏糖醇或对苯二酚。进行还原处理的时间优选5~50min。
然后,将配置的乌头酸α-β不饱和羰基化胶原溶液和巯基化胶原溶液与tg酶充分混合,利用3d打印机进样,通过计算机程序控制液体出样的速度和混合比例及出样位置,两种液体接触后固化成型,实现新型3d打印水凝胶的制备。
在本发明前述实施例提供的水凝胶制备工艺中,通过分别在胶原分子链上接枝α-β不饱和羰基化基团,在胶原分子链接枝巯基基团,通过对两种生物材料的活性结构进行修饰,利用michael加成反应和酶催化反应,以化学双交联机制来实现水凝胶的快速固化,以实现材料的3d可打印性,并通过改变经修饰后材料的浓度和tg酶的加入量来调节水凝胶的力学强度及弹性模量。本发明实施例制备的3d生物打印水凝胶固化速度快,生物相容性好、力学强度可调、在培养基中的稳定性好,生物降解速度可调,应用范围大。其与目前的紫外光固化水凝胶、ph响应水凝胶、温敏型水凝胶、离子响应水凝胶等相比,既提高了固化速度,也改善了水凝胶的机械强度和弹性,使得3d打印的水凝胶具备良好的支撑性和保真性,防止胶体在短时间内坍塌和极大程度的形变。
本发明实施例提供的制备工艺简单,能够有效地制备得到具有前述固化速度快、生物相容性好、机械强度可调、在培养基中的稳定性好以及生物降解速度可调等特点的双交联胶原水凝胶。
此外,本发明实施例还提供了前述的巯基化胶原,其是一种新的胶原巯基化改性衍生物,该类衍生物的支链结构及化学性质可根据巯基化合物调节。
进一步地,本发明实施例还提供前述的乌头酸修饰胶原,其是一种新的巯基与α-β不饱和结构接枝的产物,该产物在生物医学及组织工程领域具有重要用途。
本发明实施例的另一个方面还提供了所述双交联胶原水凝胶在制作生物医疗材料、组织工程材料或3d打印材料中的应用。
为使本发明实施例的目的、技术方案和优点更加清楚,下面将对本发明实施例中的技术方案进行清楚、完整地描述。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
实施例1本实施例提供的水凝胶的制备方法包括如下步骤:
(1)准确称取胶原1.5g,溶于质量分数为0.01%的乙酸中。在磁力搅拌器作用下基本溶解均匀后,置于超声振荡器中超声30min。准确称取乌头酸1.8g,加入5ml丙酮使其完全溶解。将溶解后的乌头酸匀速缓慢的加入胶原乙酸溶液中,在磁力搅拌器下搅拌20min左右使其充分混匀。将混合均匀的液体转入150ml圆底烧瓶中,置于集热式磁力搅拌器中,设置温度35℃,恒温加热反应2h。反应停止后透析三天,每隔5h换一次透析液。冷冻干燥三天得到col-taa产物,即乌头酸修饰胶原。
化学反应式:
(2)准确称取二巯基丁二酸1.5g,加入到氢氧化钠溶液中进行溶解。称取1.2gedc和300mgnhs同时加入上述溶液中,充分混合后,用氢氧化钠溶液调节混合溶液ph值至5,置磁力搅拌器上搅拌10min。准确称取1.5g胶原,溶于质量分数为0.01%的乙酸溶液中,将胶原溶液缓慢均匀倒入上述混合液,搅拌使混合完全。将上述混合液转移至250ml圆底烧瓶中,置于集热式磁力搅拌器中,设置温度30℃,恒温加热反应4h。反应停止后透析三天,每隔5h换一次透析液。冷冻干燥三天得到胶原巯基产物col-sh,即巯基化胶原。
化学反应式:
准确称取col-taa60mg,用质量分数1%的乙酸溶液溶解,依次进行超声震荡、氮气除气泡,配成浓度为10mg/ml的溶液。再准确称取col-sh60mg,用氢氧化钠溶液进行溶解,再进行超声震荡、氮气除气泡,配成浓度为10mg/ml的溶液,用zn处理10min,震荡混合均匀。将配好的col-taa溶液和col-sh溶液与tg酶的加入量1u/ml-6u/ml充分混合,在3d打印机上由进样系统进样,计算机程序控制出样速度和打印模式,均匀混合制备得到3d生物打印水凝胶。
实施例2本实施例提供的水凝胶的制备方法包括如下步骤:
(1)准确称取胶原1.5g,溶于质量分数为30%的乙酸中。在磁力搅拌器作用下基本溶解均匀后,置于超声振荡器中超声35min。准确称取乌头酸4.5g,加入15ml丙酮使其完全溶解。将溶解后的乌头酸匀速缓慢的加入胶原乙酸溶液中,在磁力搅拌器下搅拌30min使其充分混匀。将混合均匀的液体转入150ml圆底烧瓶中,置于集热式磁力搅拌器中,设置温度35℃,恒温加热反应10h。反应停止后透析两天,每隔4h换一次透析液。冷冻干燥两天得到col-taa产物。
化学反应式:
(2)准确称取二巯基丁二酸1.5g,加入到氢氧化钠溶液中进行溶解。称取1.2gedc和300mgnhs同时加入上述溶液中,充分混合后,用氢氧化钠溶液调节混合溶液ph至6.5左右,置磁力搅拌器上搅拌10min。准确称取1.5g胶原,溶于质量分数为10%的乙酸溶液中,将胶原溶液缓慢均匀倒入上述混合液,搅拌使混合完全。将上述混合液转移至250ml圆底烧瓶中,置于集热式磁力搅拌器中,设置温度20℃,恒温加热反应4h。反应停止后透析三天,每隔5h换一次透析液。冷冻干燥三天得到胶原巯基产物。
化学反应式:
(3)准确称取col-taa60mg,用质量分数为10%的乙酸溶液溶解,依次进行超声震荡、氮气除气泡,配成浓度为10mg/ml的溶液。再准确称取col-sh60mg,用氢氧化钠溶液进行溶解,再进行超声震荡、氮气除气泡,配成浓度为10mg/ml的溶液,用zn处理15min,震荡混合均匀。将配好的col-taa溶液和col-sh溶液与tg酶充分混合,在3d打印机上由进样系统进样,计算机程序控制出样速度和打印模式,均匀混合制备得到3d生物打印水凝胶。
实施例3本实施例提供的水凝胶的制备方法包括如下步骤:
(1)准确称取胶原1.5g,溶于质量分数30%的乙酸中。在磁力搅拌器作用下基本溶解均匀后,置于超声振荡器中超声150min。准确称取乌头酸5.4g,加入20ml双蒸水使其完全溶解。将溶解后的乌头酸匀速缓慢的加入胶原乙酸溶液中,在磁力搅拌器下搅拌150min使其充分混匀。将混合均匀的液体转入150ml圆底烧瓶中,置于集热式磁力搅拌器中,设置温度35℃,恒温加热反应10h。反应停止后透析三天,每隔5h换一次透析液。冷冻干燥三天得到col-taa产物。
化学反应式:
(2)准确称取1.5g巯基丁二酸,加入一定量双蒸水完全溶解。称取1.2gedc和300mgnhs同时加入上述溶液中,充分混合后,用氢氧化钠溶液调节混合溶液ph至5左右,置磁力搅拌器上搅拌10min。准确称取1.5g胶原,溶于0.1%质量分数的乙酸溶液,将胶原溶液缓慢均匀倒入上述混合液,搅拌使混合完全。将上述混合液转移至250ml圆底烧瓶中,置于集热式磁力搅拌器中,设置温度36℃,恒温加热反应4h。反应停止后透析三天,每隔5h换一次透析液。冷冻干燥三天得到col-sh产物。
化学反应式:
(3)准确称取col-taa240mg,用质量分数为1%的乙酸溶液溶解,超声震荡,氮气除气泡,配成浓度为100mg/ml的溶液。准确称取col-sh240mg,用氢氧化钠溶液溶解,超声震荡,氮气除气泡,配成浓度为100mg/ml的溶液,用还原剂二硫苏糖醇处理100min,震荡混合均匀。将配好的col-taa和col-sh溶液与tg酶充分混合,在3d打印机上由进样系统进样,计算机程序控制出样速度和打印模式,混合均匀制备得到3d生物打印水凝胶。
实施例4本实施例提供的水凝胶的制备方法包括如下步骤:
(1)准确称取胶原1.5g,溶于质量分数15%的乙酸中。在磁力搅拌器作用下基本溶解均匀后,置于超声振荡器中超声150min。准确称取乌头酸1.2g,加入6ml丙酮使其完全溶解。将溶解后的乌头酸匀速缓慢的加入胶原乙酸溶液中,在磁力搅拌器下搅拌40min使其充分混匀。将混合均匀的液体转入150ml圆底烧瓶中,置于集热式磁力搅拌器中,设置温度30℃,恒温加热反应6h。反应停止后透析三天,每隔4h换一次透析液。冷冻干燥三天得到col-taa产物。
化学反应式:
(2)准确称取1.5g巯基丁二酸,加入一定量双蒸水完全溶解。称取1.2gedc和300mgnhs同时加入上述溶液中,充分混合后,用氢氧化钠溶液调节混合溶液ph至6,置磁力搅拌器上搅拌8min。准确称取1.5g胶原,溶于2%质量分数的乙酸溶液,将胶原溶液缓慢均匀倒入上述混合液,搅拌使混合完全。将上述混合液转移至250ml圆底烧瓶中,置于集热式磁力搅拌器中,设置温度15℃,恒温加热反应4h。反应停止后透析三天,每隔4h换一次透析液。冷冻干燥三天得到col-sh产物。
化学反应式:
(3)准确称取col-taa240mg,用质量分数为1%的乙酸溶液溶解,超声震荡,氮气除气泡,配成浓度为100mg/ml的溶液。准确称取col-sh240mg,用氢氧化钠溶液溶解,超声震荡,氮气除气泡,配成浓度为100mg/ml的溶液,用还原剂对苯二酚处理100min,震荡混合均匀。将配好的col-taa和col-sh溶液与tg酶充分混合,在3d打印机上由进样系统进样,计算机程序控制出样速度和打印模式,混合均匀制备得到3d生物打印水凝胶。
实施例5本实施例提供的水凝胶的制备方法包括如下步骤:
(1)准确称取胶原1.5g,溶于质量分数15%的乙酸中。在磁力搅拌器作用下溶解均匀后,置于超声振荡器中超声30min。准确称取乌头酸3.6g,加入20ml丙酮使其完全溶解。将溶解后的乌头酸匀速缓慢的加入胶原乙酸溶液中,在磁力搅拌器下搅拌80min使其充分混匀。将混合均匀的液体转入150ml圆底烧瓶中,置于集热式磁力搅拌器中,设置温度30℃,恒温加热反应5h。反应停止后透析三天,每隔3h换一次透析液。冷冻干燥4天左右得到col-taa产物。
化学反应式:
(2)准确称取1.5g胶原,溶于质量分数为1%的乙酸溶液中,称取1.2gedc和300mgnhs同时加入上述溶液中,充分混合后,用氢氧化钠溶液调节混合溶液ph至6左右,置磁力搅拌器上搅拌90min。准确称取5.3g半胱氨酸乙酯,加入150ml双蒸水完全溶解,将半胱氨酸乙酯溶液缓慢均匀倒入上述混合液,搅拌使混合完全。将上述混合液转移至250ml圆底烧瓶中,置于集热式磁力搅拌器中,设置温度20℃,恒温加热反应8h。反应停止后透析三天,每隔4h换一次透析液。冷冻干燥三天左右得到col-sh产物。
化学反应式:
(3)准确称取col-taa120mg,用质量分数为1%的乙酸溶液溶解,超声震荡,氮气除气泡,配成浓度为50mg/ml的溶液。准确称取col-sh120mg,用氢氧化钠溶液溶解,超声震荡,氮气除气泡,配成浓度为50mg/ml的溶液,用还原剂处理50min左右,震荡混合均匀。将配好的col-taa和col-sh溶液与tg酶充分混合,在3d打印机上由进样系统进样,计算机程序控制出样速度和打印模式,混合均匀制备得到3d生物打印水凝胶。
实施例6本实施例提供的水凝胶的制备方法包括如下步骤:
(1)准确称取胶原1.5g,溶于质量分数15%的乙酸中。在磁力搅拌器作用下溶解均匀后,置于超声振荡器中超声30min。准确称取乌头酸3.6g,加入20ml丁酮使其完全溶解。将溶解后的乌头酸匀速缓慢的加入胶原乙酸溶液中,在磁力搅拌器下搅拌90min使其充分混匀。将混合均匀的液体转入150ml圆底烧瓶中,置于集热式磁力搅拌器中,设置温度10℃,恒温加热反应6h。反应停止后透析三天,每隔5h换一次透析液。冷冻干燥4天左右得到col-taa产物。
化学反应式:
(2)准确称取1.5g胶原,溶于质量分数为1%的乙酸溶液中,称取1.2gedc和300mgnhs同时加入上述溶液中,充分混合后,用氢氧化钠溶液调节混合溶液ph至6左右,置磁力搅拌器上搅拌90min。准确称取5.3g半胱氨酸乙酯,加入一定量双蒸水完全溶解,将半胱氨酸乙酯溶液缓慢均匀倒入上述混合液,搅拌使混合完全。将上述混合液转移至250ml圆底烧瓶中,置于集热式磁力搅拌器中,设置温度3℃,恒温加热反应8h。反应停止后透析三天,每隔3h换一次透析液。冷冻干燥三天左右得到col-sh产物。
化学反应式:
(4)准确称取col-taa120mg,用质量分数为10%的乙酸溶液溶解,超声震荡,氮气除气泡,配成浓度为50mg/ml的溶液。准确称取col-sh120mg,用氢氧化钠溶液溶解,超声震荡,氮气除气泡,配成浓度为50mg/ml的溶液,用还原剂处理50min左右,震荡混合均匀。将配好的col-taa和col-sh溶液与tg酶充分混合,在3d打印机上由进样系统进样,计算机程序控制出样速度和打印模式,混合均匀制备得到3d生物打印水凝胶。
对实施例1中的胶原、巯基化胶原和胶原以及乌头酸修饰胶原进行傅里叶红外分析图谱,分别得到图2和图3,图2中胶原与巯基化胶原红外图谱对比分析可以看出,在波数900cm-1附近,巯基化胶原有一个明显区别于胶原的弱吸收峰,说明巯基化胶原上存在c-s键,表明巯基化合物成功引入到胶原链上;图4中胶原与乌头酸接枝胶原的红外图谱对比分析可以看处,在3000cm-1附近,此处为碳碳双键上c-h键的伸缩振动峰。在1650cm-1处,此处为碳碳双键的伸缩振动峰与酰胺的吸收峰重叠,在同一波数附近变现处更强吸收。在1000cm-1附近,此处为碳碳双键上碳氢键的面外弯曲振动峰。在3400cm-1处,此处为羧基上o-h的伸缩振动峰。通过以上对比分析,表明乌头酸被成功接枝在胶原上。同时,通过扫描电镜对实施例1中的胶原巯基化衍生物、col-taa产物以及固化后的水凝胶的表面结构和表面孔径做扫描电镜(sem)分析。结果依次如图5、图6、图7、图8所示。sem分析表明固化后水凝胶表面出现很多交联后的孔洞,说明交联反应进行得很完全,进一步说明制备得到的巯基化胶原可以成功制备快速固化水凝胶。同时对巯基化胶原、乌头酸修饰的α-β不饱和羰基化胶原进行cd图谱分析,结果依次如图9、图10所示,可以看出修饰后胶原的三股螺旋结构仍然存在,保持了良好生物活性。进一步地,通过用instron力学测试机对实施例1中得到的水凝胶进行测试,使用10n传感器,置于传感器压缩基底上,根据样品的大小设置好测试参数,测试曲线出现突变后,停止压缩,系统自动得出弹性模量值。
测试参数为:长10mm,宽8mm,高5mm,压缩速率0.5mm/min。测试结果如图11所示,在压缩位移为2.87mm,压缩应力为0.133mpa时发生断裂,从而测得弹性模量为0.490mpa,因此,可以看出该水凝胶具有较好的机械强度和弹性性能。
另外,本发明其余实施例制得的水凝胶产品也都体现出优异的性能,例如都具有较为理想的机械强度和弹性性能。
综上所述,本发明实施例通过将乌头酸α-β不饱和羰基化的胶原与巯基化的胶原作为原材料,通过michael加成反应和tg酶催化反应可实现原材料由液态到固态的快速转变,大大提高了材料的可打印性,通过调节二者的浓度以及酶的加入量可实现固化后水凝胶弹性模量的调节,可扩大材料的应用范围,该方法制备的水凝胶在生物医学及组织工程领域具有重要用途,其固化速度快,生物相容性好、机械强度可调、在培养基中的稳定性好,生物降解速度可调,应用范围大。
以上所描述的实施例是本发明一部分实施例,而不是全部的实施例。本发明的实施例的详细描述并非旨在限制要求保护的本发明的范围,而是仅仅表示本发明的选定实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
- 还没有人留言评论。精彩留言会获得点赞!