与公猪精子畸形率相关的分子遗传标记及其应用和获取方法与流程
本发明涉及分子标记技术领域,特别涉及与公猪精子畸形率相关的分子遗传标记及其应用和获取方法。
背景技术:
人工受精(ai)自1954年在我国应用研究以来,至今经过数十年的技术改进和融合以及国家的支持,人工受精技术在我国越来越成熟,在大中型养殖场的应用也基本普及。人工受精的优点是不仅提高了优良公猪的利用率,还能对公猪精液品质进行适时的检测和记录。同时,通过对精液质量评定还能监控公猪健康状况和繁殖潜力,从而可优化个体遗传潜力,发挥最大繁殖能力。
在人工授精的过程中,由于精子形态不佳而导致有效精子数量降低,从而导致怀孕率下降。精子畸形率是指畸形精子占总精子的百分率。公猪的畸形精子率一般不能超过18%,否则应弃去。畸形精子指巨型精子、短小精子、断尾、断头、顶体脱落、原生质、头大、双头、双尾、折尾等精子,一般不能直线运动,受精能力较差。现今常用吉姆萨染色法测定畸形率,此种方法计数准确但费时费力。
畸形率较高,经过数月正常天气和良好的饲喂调整,再对种公猪精子进行形态观察,仍有较高的畸形率,则可认为畸形率高是由于种公猪遗传因素造成的,应予以淘汰。
因此,挖掘和利用新的预防精子畸形率基因对于猪的遗传育种有着重大意义。基于覆盖全基因组的高密度snp数据和大群体的性状表型记录,可通过全基因组关联分析技术(gwas)准确定位控制性状的候选基因。尽管该技术仍然存在一些缺陷,其已被广泛应用于人类复杂疾病候选基因挖掘和畜禽重要经济性状关键基因的定位。经典的gwas一般基于plink等软件对所有标记逐个进行单标记回归分析,继而设定一个显著阈值来筛选显著位点。这类方法往往面临计算强度大、过高估计标记效应、显著性阈值设定不合理等问题。为了进一步提高gwas的效率,新方法和软件不断被提出。其中,一步法全基因组关联分析(wssgwas)同时利用系谱、历史个体表型记录和基因型数据进行关联分析,适用于大量个体拥有表型记录而只有少量个体拥有基因型数据的情况,尤其适用于畜禽重要经济性状的全基因组关联分析。基于gblupf90软件,可轻易实现wssgwas。在不同的研究群体中,前人已经定位到了部分影响公猪精液性状的候选基因或者基因组区域。
技术实现要素:
鉴于上述内容,有必要提供与公猪精子畸形率相关的分子遗传标记及其应用和获取方法,该分子遗传标记位于猪的第3号染色体的第8647231bp位置为c>t突变;可根据该突变设计出用于扩增该分子标记的引物和鉴定分子标记的探针;进而应用于筛选具有高精子畸形率的公猪,从而将其应用于猪的人工授精;本申请利用一步法全基因组关联分析法进行分析,能有效提高筛选分子标记的准确率和效率。
为达到上述目的,本发明所采用的技术方案是:
本发明的猪基因组序列参考国际猪基因组10.2版本参考序列:
与公猪精子畸形率相关的分子遗传标记,所述的分子遗传标记位于猪的第3号染色体的第8647231核酸位点,该位点的碱基为c或t,对应位于核酸序列表seqidno.1的第101位核酸位点。(申请人命名为wu_10.2_3_8041535)
本申请还提供一种用于扩增所述分子遗传标记的引物或鉴定所述分子遗传标记的探针。
本申请还提供一种含有所述引物或探针的试剂盒。
本申请还提供一种所述分子遗传标记在检测公猪精子畸形率能力、辅助猪的人工授精、辅助选育和/或选育精子畸形率能力低的种猪中的应用。
本申请还提供一种选育或辅助选育公猪精子畸形率能力高的种猪的方法,所述方法为:提取公猪的总dna,检测公猪第3号染色体的第8647231位核苷酸位点,测出第8647231位核苷酸的序列为c或t、或c和t,确定待测猪的基因型是cc型、tt型或tc型,选择cc型基因的公猪进行下一步选种和/或育种。
进一步的,所述cc基因型为公猪第3号染色体的第8647231位核苷酸c的纯合体;tt基因型为3号染色体的第8647231位核苷酸为t的纯合体;tc基因型为猪第3号染色体的第8647231位核苷酸为c与t的杂合体。
本发明还提供了一种获取与公猪精子畸形率相关的分子遗传标记的方法,所述方法为:采集公猪的耳组织样品和/或血液为样品,提取总dna,并对dna进行质量检测,获得全基因组的snp标记基因型;采用基因比对的方法对获得的snp标记的物理位置,对于基因组位置未知的snp不用于关联分析,对所有常染色体上的snp标记,进行质量控制筛选出snp,之后对筛选出来的snp进行全基因组关联分析获得分子遗传标记,所述质量控制标准为:个体检出率≥90%;snp检出率≥90%;最小等位基因频率≥0.01;哈迪温伯格平衡的p值≥106。
进一步的,所述全基因组关联分析方法为:
为了充分利用所有表型数据和基因型数据,本发明专利采用加权的一步法全基因组关联分析法(weightedsinglestepgenome-wideassociationstudy,wssgwas)进行全基因组关联分析。该方法首先基于混合模型方程组来估计个体育种值,继而基于育种值模型与标记效应模型的等价关系将育种值转换为标记效应。本发明采用的全基因组关联分析模型如下:
y=xb+za+wp+age+intv+e
式中,y为精子畸形率观测值向量;
x,z,w为设计矩阵;
b为固定效应向量(总体均值和年-季效应);
p为个体永久环境效应,
age为公猪采精时的月龄协变量;
intv为公猪采精间隔协变量;
e为残差,
a为育种向量,
式中,a为基于系谱的亲缘关系矩阵;
a22为a中有基因型个体对应的分块矩阵;
对应上述混合模型,采用ai-reml(averageinformationrestrictedmaximumlikelihood)法估计方差组分,并通过求解混合模型方程组获得育种值。通过迭代的方式获得标记权重,主要步骤如下:
第1步:初始化(t=1),d(t)=i,g(t)=λzd(t)z',
第2步:通过ssgblup计算个体育种值;
第3步:通过公式
第4步:利用公式
第5步:利用公式
第6步:利用公式g(t+1)=λzd(t+1)z'计算亲缘关系矩阵用于下一轮迭代;
第7步:令t=t+1,并从第2步开始下一轮迭代。
上述步骤迭代三次,最终获得snp标记效应。将第三轮迭代输出的标记效应作为最终的结果。计算过程主要通过在r统计分析平台编程调用blupf90软件来实现,其中airemlf90程序用于方差组分估计,blupf90程序用于计算育种值,postgsf90用于计算标记效应。
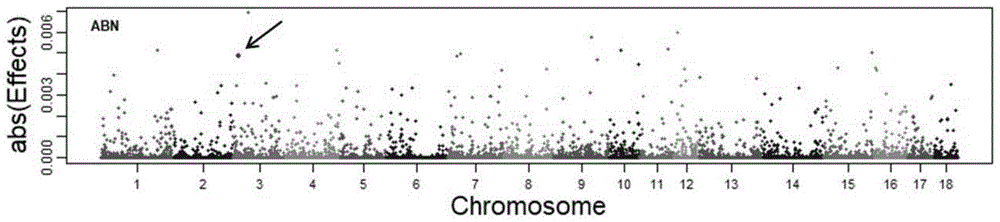
对于所有标记的效应值,取其绝对值画曼哈顿图,展示和筛选大效应的snp标记。并采用方差分析和多重比较(r统计分析平台),分析wu_10.2_3_8041535标记不同基因型群体的公猪精子畸形率差异情况。
本发明具有如下有益效果:
(1)本发明专利鉴定了一个影响公猪精子畸形率的分子标记wu_10.2_3_8041535(猪第3号染色体的第8647231位核酸位点)用于标记不同基因型公猪的精子畸形率有极显著差异;鉴定结果证明,对于wu_10.2_3_8041535标记来说,基因型cc与的畸形率低于tc型和tt型;可通过上述位点的基因型选择进行辅助公猪育种或选育,将对于wu_10.2_3_8041535的cc型公猪进行留选,并送入公猪站,可有效降低精子畸形率,有效提高公猪人工授精的成功率;同时,本申请采用一步法全基因组关联分析(wssgwas)同时利用系谱、历史个体表型记录和基因型数据进行关联分析,适用于大量个体拥有表型记录而只有少量个体拥有基因型数据的情况,尤其适用于畜禽重要经济性状的全基因组关联分析。基于gblupf90软件,可轻易实现wssgwas;本发明中发现的snp与公猪精液畸形率性状的相关性达到了极显著水平,为公猪精液畸形率性状的研究提供了新的遗传资源。
【附图说明】
图1是wu_10.2_3_8041535标记基因组位置及精子畸形率全基因组snp效应分布图。
【具体实施方式】
为使本发明的上述目的、特征和优点能够更加明显易懂,下面结合附图对本发明的具体实施方式做详细的说明。在下面的描述中阐述了很多具体细节以便于充分理解本发明。但是本发明能够以很多不同于在此描述的其它方式来实施,本领域技术人员可以在不违背本发明内涵的情况下做类似改进,因此本发明不受下面公开的具体实施的限制。
实施例1:
1、表型-系谱数据采集
本申请的基础研究群体为杜洛克公猪,全部来自广西秀博股份有限公司公猪站,完整系谱中包含12个世代5284头种猪,其中2015-2018年间记录了2693头公猪的精子畸形率性状表型数据;精子畸形率通过ultimatetmcasa(hamiltonthorneinc.,beverly,ma,usa)系统对新鲜精液进行分析获得。总共获得143114条精液性状观测值(平均每头公猪53条数据),用于表型-基因型关联分析。
2、基因分型与质量控制
采集1733头公猪的耳组织样品或者血样,提取总dna,并采用ggp50ksnp(geneseek,us)芯片进行基因分型,获得覆盖全基因组的50705个snp标记。根据最新版的猪参考基因组(sscrofa11.1),采用ncbi基因组比对程序、对所有snp标记的物理位置进行更新。基因组位置未知的snp不用于关联分析。对于所有常染色体上的snp标记,利用plink软件进行质量控制,标准为:个体检出率≥90%;snp检出率≥90%;最小等位基因频率≥0.01;哈迪温伯格平衡的p值≥106,采用beagle软件(version4.1)进行填充;基于以上质量控制标准,剩余1623头公猪和28289个snp标记用于关联分析,其中1231头公猪既有精子畸形率表型数据,也有基因型数据。
(3)统计模型
为了充分利用所有表型数据和基因型数据,本发明专利采用加权的一步法全基因组关联分析法进行全基因组关联分析。该方法首先基于混合模型方程组来估计个体育种值,继而基于育种值模型与标记效应模型的等价关系将育种值转换为标记效应。本发明采用的全基因组关联分析模型如下:
y=xb+za+wp+age+intv+e
式中,y为精子畸形率观测值向量;
x,z,w为设计矩阵;
b为固定效应向量(总体均值和年-季效应);
p为个体永久环境效应,
age为公猪采精时的月龄协变量;
intv为公猪采精间隔协变量;
e为残差,
a为育种向量,
式中,a为基于系谱的亲缘关系矩阵;
a22为a中有基因型个体对应的分块矩阵;
gω=0.9g+0.1a22,
对应上述混合模型,采用ai-reml(averageinformationrestrictedmaximumlikelihood)法估计方差组分,并通过求解混合模型方程组获得育种值。通过迭代的方式获得标记权重,主要步骤如下:
第1步:初始化(t=1),d(t)=i,g(t)=λzd(t)z',
第2步:通过ssgblup计算个体育种值;
第3步:通过公式
第4步:利用公式
第5步:利用公式
第6步:利用公式g(t+1)=λzd(t+1)z'计算亲缘关系矩阵用于下一轮迭代;
第7步:令t=t+1,并从第2步开始下一轮迭代。
上述步骤迭代三次,最终获得snp标记效应。将第三轮迭代输出的标记效应作为最终的结果。计算过程主要通过在r统计分析平台编程调用blupf90软件来实现,其中airemlf90程序用于方差组分估计,blupf90程序用于计算育种值,postgsf90用于计算标记效应。
(4)标记筛选
对于所有标记的效应值,取其绝对值画曼哈顿图,图示如图1所示;展示和筛选大效应的snp标记。并采用方差分析和多重比较(r统计分析平台),分析wu_10.2_3_8041535标记不同基因型群体公猪精子畸形率差异情况,具体如表1所示:
表1
由上表可知,cc纯合基因型的公猪精子畸形率低于tc型杂合基因畸形率,tc型杂合基因畸形率低于tt纯合基因畸形率。
实施例2:
根据上述筛选得到的基因结果,显示,本申请与公猪精子畸形率相关的分子遗传标记,所述的分子遗传标记位于猪3号染色体8647231bp位置,该位置为一个t>c突变(sscrofa10.2),对应位于核酸序列表seqidno.1的第101位核酸位点。
实施例3:
本领域技术人员很容易根据本发明的分子遗传标记设计出用于扩增该分子标记的引物或鉴定所述分子标记的探针,从而用于该遗传标记的检测,例如通过pcr扩增得到所述分子遗传标记,再通过克隆测序得到相应的序列,或者通过bsm-rflp多态性进行检测。因此,本发明还包括用于扩增所述分子遗传标记的引物或鉴定所述分子遗传标记的探针,以及含有所述引物或探针的试剂盒。
实施例4:
可应用本申请的分子遗传标记辅助进行公猪精子畸形率能力的检测,具体方法为:提取公猪的基因组dna,设计引物扩增如序列表seqidno.1的基因片段(位于猪第15号染色体上),并检测其第101位点的基因为c或t;根据该位点基因型判断待测猪是cc型、tc型或tt型;然后根据已知的验证结果(表1)得出:cc纯合基因型的公猪精子畸形率低于tc型杂合基因畸形率,tc型杂合基因畸形率低于tt纯合基因畸形率。
实施例5:
可应用本申请的分子遗传标记辅助进行公猪的人工授精工作,具体方法为:提取公猪的基因组dna,设计引物扩增如序列表seqidno.1的基因片段,并检测其第101位点的基因为c或t;根据该位点基因型判断待测猪是cc型、tc型或tt型;选择cc型公猪进入公猪站进行人工授精。
实施例6:
可应用本申请的分子遗传标记辅助进行公猪的育种或辅助育种工作,具体方法为:提取公猪的基因组dna,设计引物扩增如序列表seqidno.1的基因片段,并检测其第101位点的基因为c或t;根据该位点基因型判断待测猪是cc型、tc型或tt型;根据育种需求,选择cc型、tc型或tt型的公猪进行留种或配种;其中,cc纯合基因型的公猪精子畸形率低于tc型杂合基因畸形率,tc型杂合基因畸形率低于tt纯合基因畸形率。
综上所述,使用本申请的方法能简单、高效、准确的获取与公猪精子畸形率相关的分子遗传标记,根据该突变可设计出用于扩增该分子标记的引物和鉴定分子标记的探针;快速筛选出具有高精子畸形率能力的公猪,从而将其应用于猪的人工授精;快速筛选出具有高精子畸形率能力的公猪,本申请利用一步法全基因组关联分析法进行分析,能有效提高筛选分子标记的准确率和效率。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明的保护范围应以所附权利要求为准。
序列表
<110>广西扬翔农牧有限责任公司
<120>与公猪精子畸形率相关的分子遗传标记及其应用和获取方法
<160>1
<170>siposequencelisting1.0
<210>1
<211>201
<212>dna
<213>猪属(susscrofa)
<220>
<221>misc_feature
<222>(101)..(101)
<223>niscort
<400>1
gtaggacagtctgtcacaaccatcagcaccaagcgtctcttctctctgccctgagagaca60
catatgaacttctgatatgttcacttatgcgttcgacaaangttgattgggtgccttcaa120
ttctcttggccttggggtcaccatgaaaagggtgtgtccttgctctcatgctgttagatt180
ctaaggggcacatcaggtctt201
- 还没有人留言评论。精彩留言会获得点赞!