一种引导PD1基因切割实现外源序列高效整合的sgRNA的制作方法
一种引导pd1基因切割实现外源序列高效整合的sgrna
技术领域
[0001]
本发明涉及一种核苷酸,尤其涉及一种sgrna,该sgrna能引导pd1基因座切割,从而实现外源序列在该位点的高效整合。
背景技术:
[0002]
嵌合抗原受体t细胞技术(chimeric antigen receptor t-cell,car-t)是近年来兴起的一种新型过继免疫疗法。它将患者t细胞在体外进行基因改造,经过一定扩增后回输至患者体内,从而实现肿瘤的靶向杀伤。car-t结构主要包括胞外特异性识别肿瘤特定抗原的单链抗体、跨膜结构域、胞内t细胞活化的串联信号结构域(目前使用较多的是cd28-cd3z和4-1bb-cd3z)。自从2013年《科学》杂志将肿瘤免疫治疗技术评选为年度十大科技突破,该领域的研究得到了蓬勃发展。目前,已有两款car-t产品被美国食品药品监督管理局批准用于血液肿瘤的临床治疗,标志着car-t技术的巨大成功。但是,作为新兴的技术,car-t技术在各方面依然面临着很多挑战,有巨大的改进空间。
[0003]
crispr/cas9系统是来源于古细菌和细菌的一种抵御质粒、噬菌体等外源dna片段入侵的获得性免疫机制。该系统主要由crispr序列和编码cas蛋白的基因座发挥功能,目前最常用的是-型家族的cas9核酸酶,这一系统只需要单一的效应蛋白就可发挥作用。当外源dna入侵细菌时,-型crispr系统首先将入侵dna整合到crispr重复序列之间。随后,包含入侵dna序列的crispr rna(crrna)被转录和加工出来。此后,反式激活crrna(transactivating crispr rna,tracrrna)与crrna结合,并最终与cas9蛋白形成复合物。最后,cas9蛋白通过其hnh和ruvc样结构域发挥核酸内切酶作用引发dna双链断裂。dna的双链断裂会引发损伤修复机制,从而通过同源重组等方式实现特定基因的编辑。迄今,crispr/cas9技术已经在很多领域得以成功应用,显示出广阔的前景。
[0004]
pd-1是表达在t细胞表面的一种重要的免疫抑制跨膜蛋白,它有两个配体pd-l1和pd-l2。研究发现肿瘤细胞能高表达pd-l1激活pdl1/pd1通路,使pd-1的胞内结构域发生磷酸化,招募酪氨酸磷酸酶shp-2,从而减少tcr信号通路的活化,抑制t细胞的激活。临床研究表明阻断pdl1/pd1通路可以解除t细胞的抑制作用,起到杀伤肿瘤的效果。目前已有多款阻断pdl1/pd1通路的抗体药物被美国食品药品监督管理局批准用于非小细胞肺癌、黑色素瘤等多种恶性肿瘤的治疗,体现出良好的疗效,标志着pd1免疫检查点疗法的巨大成功。
[0005]
为了提高car-t细胞的抗肿瘤效果,本发明利用基因编辑技术对pd1基因座特定位点dna进行了切割,并同时导入外源性的目的序列,提高了car-t技术的应用空间。
技术实现要素:
[0006]
本发明提供了一种针对pd1基因进行基因编辑的方法。
[0007]
本发明中,“基因编辑”是指在细胞的基因的靶点处进行的取代、缺失和/或插入。在一个实施方式,基因编辑可以通过将一个或多个天然的或人工改造的核酸酶引入到细胞中以在细胞的靶点处产生dna突变;在其他的实施方式中,可以提供外源的供体修复模板。
[0008]
本发明中,可以采用本领域常规的技术将核酸酶引入到细胞中,例如,可以采用载体转化、显微注射、转染、脂质转染、热休克、电穿孔、转导、基因枪、显微注射、deae-葡聚糖介导的转移等将核酸酶引入到细胞中;在优选的实施方式中,所述细胞可以为t细胞;所述引入的方式可以采用电穿孔。
[0009]“t细胞”或“t淋巴细胞”是本领域公认的包括胸腺细胞、初始t淋巴细胞、未成熟t淋巴细胞、成熟t淋巴细胞、静息t淋巴细胞或活化t淋巴细胞。t细胞可以是t辅助(th)细胞,例如t辅助1(th1)细胞或t辅助2(th2)细胞。t细胞可以是辅助t细胞(htl;cd4+t细胞)、cd4+t细胞、细胞毒性t细胞(ctl;cd8+t细胞)、肿瘤浸润细胞毒性t细胞(til;cd8+t细胞)、cd4
+
cd8
+
t细胞、cd4-cd8-t细胞或任何其它t细胞亚群。在一个实施方式中,t细胞可以是nkt细胞。在优选的实施方式中,t细胞被修饰以表达car。
[0010]
本发明中,可以使用核酸酶编辑细胞中的pd1基因,所述核酸酶可以结合和切割pd1基因上的靶点序列。所述核酸酶可以用于在靶多核苷酸序列中引入双链断裂,所述双链断裂可以在不存在多核苷酸模板例如供体修复模板的情况下通过非同源性末端接合(nhej)进行修复;或者在存在供体修复模板的情况下通过同源定向修复(hdr)即同源重组进行修复。
[0011]
本发明中,核酸酶是指包括一个或多个dna结合结构域和一个或多个dna切割结构域的核酸酶。本发明中的核酸酶可以是由天然存在的核酸酶或由人工改造的核酸酶设计和/或修饰而来。本发明的核酸酶包括归巢核酸内切酶、megatals、转录激活子样效应因子核酸酶(talen)、锌指核酸酶(zfn)以及成簇的规律间隔的短回文重复序列crispr/cas核酸酶系统。在优选的实施方式中,本发明的核酸酶包括crispr/cas核酸酶系统。
[0012]
本发明中,crispr/cas核酸酶系统包括cas核酸酶和将cas核酸酶募集到靶位点的一个或多个rna,例如反式活化crna(tracrrna)和crispr rna(crrna)或单向导rna(sgrna)。crrna和tracrrna可以设计“单向导rna”或“sgrna”的一个多核苷酸序列。
[0013]
在一个实施方式中,cas核酸酶包括双链dna核酸内切酶活性或切口酶活性,并且与crrna和tracrrna或sgrna形成靶络合物以便对位于pd1基因座内的原型间隔靶序列进行位点特异性dna识别和和位点特异性切割。原型间隔基序邻接短的原型间隔相邻基序(pam),其在募集cas/rna络合物中起作用。cas多肽识别对cas多肽具有特异性的pam基序。
[0014]
本发明中,cas酶是i型或iii型cas酶,优选ii型cas酶。本发明中的ii型cas酶可以是任何cas酶,包括但不限于cas9、cas3、cas8a、cas8b、cas10d、cse1、csy1、csn2、cas4、cas10、csm2、cmr5、fok1以及本领域已知的其它核酸酶和其任意组合。
[0015]
在优选的实施方式中,本发明的cas酶为cas9,更优选的,cas9酶来源于肺炎链球菌、化脓性链球菌或嗜热链球菌的cas9,此处,来源于肺炎链球菌、化脓性链球菌或嗜热链球菌应当理解为来自或衍生自肺炎链球菌、化脓性链球菌或嗜热链球菌的cas9。对于衍生的酶,意指与野生型酶具有高度序列同源性的含义,例如,在某些位点上已经被突变或修饰。
[0016]
在一些实施方式中,cas酶包含一个或多个选自下组的突变:d10a、e762a、h840a、n854a、n863a或d986a和/或其他的一个或多个在cas酶的ruvc1或hnh结构域的突变。在优选的实施方式中,所述cas酶突变包括:酿脓链球菌(d10a)、嗜热链球菌(d9a)、齿垢密螺旋体(d13a)、脑膜炎双球菌(d16a)、酿脓链球菌(d839a、h840a或n863a)、嗜热链球菌(d598a、
h599a或n622a)、齿垢密螺旋体(d878a、h879a或n902a)、以及脑膜炎双球菌(d587a、h588a或n611a)。
[0017]
在一个实施方式中,cas酶具有一个或多个在催化结构域中的突变,其中在转录时,该tracr配对序列杂交到该tracr序列上,并且该指导序列指导crispr复合物与该靶序列的序列特异性结合,并且其中该酶进一步包含一个功能结构域。
[0018]
在其他的实施方式中,适合的cas9多肽还可以从以下物种中获得:屎肠球菌、肠球菌、无害学斯特菌、单核细胞增生李斯特氏菌、斯氏利斯特菌、伊氏利斯特菌、无乳链球菌、咽峡炎链球菌、牛链球菌、停乳链球菌、马肠链球菌、解没食子酸链球菌、猕猴链环菌、变异链球菌、假链球菌、酿脓链球菌、嗜热链球菌、格氏链球菌、婴儿链球菌、马链球菌、缓症链球菌、巴氏链球菌、猪链球菌、前庭链球菌、血链球菌、道恩链球菌、杆状奈瑟菌、灰色奈瑟菌、浅黄色奈瑟菌、乳糖奈瑟菌、脑膜炎奈瑟菌、浅黄奈瑟菌、短乳杆菌、布氏乳杆菌、干酪乳杆菌、乳酸菌、发酵乳杆菌、加氏乳杆菌、詹氏乳杆菌、约氏乳杆菌、鼠李糖乳杆菌、瘤胃乳酸杆菌、唾液乳杆菌、旧金山乳杆菌、拥挤棒杆菌、白喉杆菌、马氏棒状杆菌、空肠弯曲杆菌、产气荚膜梭状芽胞杆菌、文氏密螺旋体、蚀疮溃疡密螺旋体以及齿垢密螺旋体。
[0019]
在其他的实施方式中,所述核酸酶还可以选自cpf1,cpf1可以从包括但不限于以下的细菌物种获得:弗朗西斯菌属、氨基酸球菌属、普鲁氏杆菌属、毛螺科菌属等。
[0020]
本发明中引导核酸序列sgrna特异于基因并且靶向该基因进行cas核酸内切酶-诱导的双链断裂。引导核酸序列的序列可以在基因的基因座内。在一个实施方式中,引导核酸序列的长度是至少10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、30、31、32、33、34、35、36、37、38、39、40个或更多个核苷酸。
[0021]
引导核酸序列可以特异于任何基因,本发明中,sgrna特异于pd1基因。
[0022]
本发明中,供体修复模板包括一个或多个同源臂。“同源臂”是指供体模板中与通过核酸酶在靶点处引入的dna断裂两侧的dna序列完全相同或基本完全相同的核酸序列。
[0023]
在一个实施方式中,供体模板包括5
’
同源臂,所述5
’
同源臂包括与dna断裂位点的5
’
处的dna序列完全相同或几乎完全相同的核酸。在其他的实施方式中,供体模板包括3
’
同源臂,所述3
’
同源臂包括与dna断裂位点的3
’
处的dna序列完全相同或几乎完全相同的核酸。在优选的实施方式中,供体模板包括5
’
同源臂和3
’
同源臂。
[0024]
本发明的同源臂具有合适的长度,包括但不限于:100bp到3000bp,在具体的实施方式中,同源臂的长度可以选自100bp、200bp、300bp、400bp、500bp、600bp、700bp、800bp、900bp、1000bp、1100bp、1200bp、1300bp、1400bp、1500bp、1600bp、1700bp、1800bp、1900bp、2000bp、2100bp、2200bp、2300bp、2400bp、2500bp、2600bp、2700bp、2800bp、2900bp或3000bp或更长的同源臂,以及包含所有中间长度的同源臂。
[0025]
在优选的实施方式中,供体模板包括5
’
同源臂、car以及3
’
同源臂。
[0026]
在优选的实施方式中,通过包含嵌合抗原受体(car)的外源供体模板将car插入到pd1的基因座中。
[0027]
car是将针对靶抗原(例如,肿瘤抗原)的基于抗体的特异性与t细胞受体活化细胞内结构域组合以产生嵌合蛋白的分子,所述嵌合蛋白展现出特异性抗肿瘤细胞免疫活性。
[0028]
本发明中,car包括结合到特异性靶抗原的细胞外结构域(也被称为结合结构域或抗原特异性结合结构域)、跨膜结构域以及细胞内信号传导结构域。
[0029]
在一个实施方式中,car包括特异性结合到在肿瘤细胞上表达的靶多肽例如靶抗原的细胞外结合结构域。结合结构域可以包括拥有特异性识别和结合到生物分子(例如,细胞表面受体或肿瘤蛋白、脂质、多糖或其它细胞表面靶分子或其组分)的能力的任何蛋白质、多肽、寡肽或肽。结合结构域包含兴趣生物分子的任何天然存在的、合成的、半合成的或重组产生的结合配偶体。
[0030]
进一步的,细胞外结合结构域包括抗体或其抗原结合片段。
[0031]
抗体是指作为包括至少轻链或重链免疫球蛋白可变区的多肽的结合剂,所述轻链或重链免疫球蛋白可变区特异性识别和结合靶抗原的表位,如肽、脂质、多糖或含有抗原决定簇的核酸,如由免疫细胞识别的核酸。抗体包含抗原结合片段,例如,骆驼ig(骆驼科抗体或其vhh片段)、ig nar、fab片段、fab
′
片段、f(ab)
′
2片段、f(ab)
′
3片段、fv、单链fv抗体(“scfv”)、bis-scfv、(scfv)2、微抗体、双抗体、三抗体、四抗体、二硫键稳定的fv蛋白(“dsfv”)以及单结构域抗体(sdab、纳米抗体)或其其它抗体片段。在优选的实施方式中,结合结构域是scfv。
[0032]
在一个实施方式中,所述car包括结合选自以下任一或任意几个抗原的细胞外结构域:α叶酸受体、5t4、αvβ6整联蛋白、bcma、b7-h3、b7-h6、caix、cd16、cd19、cd20、cd22、cd30、cd33、cd44、cd44v6、cd44v7/8、cd70、cd79a、cd79b、cd123、cd138、cd171、cea、cspg4、egfr、包含erbb2(her2)的egfr家族、egfrviii、egp2、egp40、epcam、epha2、epcam、fap、胎儿achr、frα、gd2、gd3、磷脂酰肌醇蛋白聚糖-3(gpc3)、hla-a1+mage1、hla-a2+mage1、hla-a3+mage1、hla-a1+ny-eso-1、hla-a2+ny-eso-1、hla-a3+ny-eso-1、il-11rα、il-13rα2、lambda、lewis-y、kappa、间皮素、muc1、muc16、ncam、nkg2d配体、ny-eso-1、prame、psca、psma、ror1、ssx、存活蛋白、tag72、tem、vegfr2以及wt-1。
[0033]
在一些实施方式中,car在各个结构域之间包括接头残基。“可变区连接序列”是将重链可变区连接到轻链可变区,并且提供与所述两个子结合结构域的相互作用相容的间隔功能的氨基酸序列,使得所得多肽保留与包括相同轻链可变区和重链可变区的抗体相同的靶分子的特异性结合亲和力。在具体实施例中,接头分离一个或多个重链可变结构域或轻链可变结构域、铰链结构域、跨膜结构域、共刺激结构域和/或初级信号传导结构域。在具体实施例中,car包括一个、两个,三个、四个或五个或更多个接头。在具体实施例中,接头的长度为约1个氨基酸到约25个氨基酸、约5个氨基酸到约20个氨基酸、或约10个氨基酸到约20个氨基酸或任何中间长度的氨基酸。在一些实施例中,接头的长度为1个、2个、3个、4个、5个、6个、7个、8个、9个、10个、11个、12个、13个、14个、15个、16个、17个、18个、19个、20个、21个、22个、23个、24个、25或更多个氨基酸。
[0034]
在一个实施方式中,car的结合结构域之后是一个或多个“间隔结构域”,所述间隔结构域是指使抗原结合结构域移动远离效应细胞表面以实现适当的细胞/细胞接触、抗原结合和活化的区。间隔结构域可以源自天然、合成、半合成或重组来源。在某些实施例中,间隔结构域是免疫球蛋白的一部分,包含但不限于一个或多个重链恒定区,例如ch2和ch3。间隔结构域可以包含天然存在的免疫球蛋白铰链区或改变的免疫球蛋白铰链区的氨基酸序列。在一个实施方式中,间隔结构域包括igg1、igg4或igd的ch2和ch3。
[0035]
car还包括跨膜结构域,跨膜结构域可以源自天然、合成、半合成或重组来源的结构域。在一个实施方式中,跨膜结构域可以包括或衍生自以下组的一个或多个跨膜区:t细
胞受体的α或β链、cd3δ、cd3ε、cd3γ、cd3ζ、cd4、cd5、cd8α、cd9、cd16、cd22、cd27、cd28、cd33、cd37、cd45、cd64、cd80、cd86、cd134、cd137、cd152、cd154以及pd-1。
[0036]
car还包括细胞内信号传导结构域,细胞内信号传导结构域将有效car结合靶抗原的信息转导到免疫效应细细胞内部以引发效应细胞功能,例如,活化、细胞因子产生、增殖和细胞毒活性,包含向car结合的靶细胞释放细胞毒性因子或使用与细胞外car结构域进行的抗原结合引发的其它细胞应答。
[0037]
细胞内信号传导结构域包括一个或多个“共刺激信号传导结构域”和一个“初级信号传导结构域”;适合的初级信号传导结构域包括fcrγ、fcrβ、cd3γ、cd3δ、cd3ε、cd3ζ、cd22、cd79a、cd79b以及cd66d。
[0038]“共刺激信号传导结构域”或“共刺激结构域”是指共刺激分子的细胞内信号传导结构域。适合的共刺激分子包括tlr1、tlr2、tlr3、tlr4、tlr5、tlr6、tlr7、tlr8、tlr9、tlr10、card11、cd2、cd7、cd27、cd28、cd30、cd40、cd54(icam)、cd83、cd134(ox40)、cd137(4-1bb)、cd278(icos)、dap10、lat、nkd2c、slp76、trim以及zap70。
[0039]
发明详述:
[0040]
一方面,本发明提供了一种针对细胞内pd1基因进行基因编辑的方法,所述方法包括利用核酸酶和sgrna对pd1基因进行基因编辑的步骤,所述sgrna引导核酸酶对pd1基因进行切割并形成断裂位点。
[0041]
进一步的,所述sgrna靶向pd1的靶向序列包含seq id no.1-6中的一条或任意几条所示的序列。
[0042]
进一步的,所述核酸酶选自cas9、cas3、cas8a、cas8b、cas10d、cse1、csy1、csn2、cas4、cas10、csm2、cmr5、fok1、cpf1中的一种或任意几种。
[0043]
在优选的实施方式中,所述核酸酶为cas9;优选的,所述cas9选自来源于肺炎链球菌、化脓性链球菌或嗜热链球菌的cas9。
[0044]
在一个实施方式中,所述sgrna还包括碱基的化学修饰。在优选的实施方式中,所述sgrna包括5
’
末端第1-n位碱基的任意一个、任意几个或任意连续几个碱基的化学修饰,和/或3
’
末端第1至n位碱基的任意一个、任意几个或任意连续几个碱基的化学修饰;所述n选自2、3、4、5、6、7、8、9或10。优选的,sgrna包括5
’
末端一个、两个、三个、四个或五个碱基的化学修饰,和/或3
’
末端一个、两个、三个、四个或五个碱基的化学修饰。例如,在sgrna的5
’
末端第1位碱基、第2位碱基、第3位碱基、第4位碱基、第5位碱基或第1-2位碱基、第1-3位碱基、第1-4位碱基、第1-5位碱基进行化学修饰;和/或,在sgrna的3
’
末端第1位碱基、第2位碱基、第3位碱基、第4位碱基、第5位个碱基或第1-2位碱基、第1-3位碱基、第1-4位碱基、第1-5位碱基进行化学修饰。在优选的实施方式中,所述化学修饰为甲基化修饰、甲氧基修饰、氟化修饰或硫代修饰中的一种或任意几种。
[0045]
所述sgrna的靶向序列包含seq id no.4-5的一种或任意几条。
[0046]
更优选的,在靶向序列的5
’
端前三个碱基进行2-甲氧基修饰和/或硫代修饰;更优选的,在靶向序列的5
’
端前三个碱基均进行2-甲氧基修饰和硫代修饰。
[0047]
进一步的,所述方法还包括提供供体修复模板的步骤。
[0048]
进一步的,所述供体修复模板包括同源臂,所述同源臂包括5
’
同源臂和/或3
’
同源臂;优选的,所述5
’
同源臂的长度为100bp到3000bp,所述3
’
同源臂的长度为100bp到
3000bp。
[0049]
进一步的,所述供体修复模板还包括外源序列。
[0050]
进一步的,所述供体修复模板还包括嵌合抗原受体(car)。
[0051]
进一步的,所述car包括结合到特异性靶抗原的细胞外结构域、跨膜结构域以及细胞内信号传导结构域。
[0052]
进一步的,所述细胞外结构域靶向的抗原选自以下一种或任意几种:α叶酸受体、5t4、αvβ6整联蛋白、bcma、b7-h3、b7-h6、caix、cd16、cd19、cd20、cd22、cd30、cd33、cd44、cd44v6、cd44v7/8、cd70、cd79a、cd79b、cd123、cd138、cd171、cea、cspg4、egfr、包含erbb2(her2)的egfr家族、egfrviii、egp2、egp40、epcam、epha2、epcam、fap、胎儿achr、frα、gd2、gd3、磷脂酰肌醇蛋白聚糖-3(gpc3)、hla-a1+mage1、hla-a2+mage1、hla-a3+mage1、hla-a1+ny-eso-1、hla-a2+ny-eso-1、hla-a3+ny-eso-1、il-11rα、il-13rα2、lambda、lewis-y、kappa、间皮素、muc1、muc16、ncam、nkg2d配体、ny-eso-1、prame、psca、psma、ror1、ssx、存活蛋白、tag72、tem、vegfr2以及wt-1。
[0053]
进一步的,所述跨膜结构域选自以下任意一个或任意几个跨膜区:t细胞受体的α或β链、cd3δ、cd3ε、cd3γ、cd3ζ、cd4、cd5、cd8α、cd9、cd16、cd22、cd27、cd28、cd33、cd37、cd45、cd64、cd80、cd86、cd134、cd137、cd152、cd154。
[0054]
进一步的,所述细胞内信号传导结构域包括共刺激信号传导结构域和/或初级信号传导结构域。
[0055]
在优选的实施方式中,所述细胞为t细胞。
[0056]
进一步的,核酸酶、sgrna和供体修复模板引入细胞的方式包括:载体转化、显微注射、转染、脂质转染、热休克、电穿孔、转导、基因枪;此处的载体应当理解为将核酸酶、sgrna或供体修复模板重组到载体上,然后将载体转化入细胞中;优选的,可以采用电穿孔的方式。更优选的,将核酸酶和sgrna形成复合物,或者将核酸酶、sgrna和供体修复模板形成复合物,再将复合物通过电穿孔的方式引入到细胞中。
[0057]
另一方面,本发明还提供了用于对细胞内pd1基因进行基因编辑的sgrna。
[0058]
另一方面,本发明还提供了上述sgrna在对细胞内pd1基因进行基因编辑的应用。
[0059]
另一方面,本发明还提供了上述基因编辑方法制备得到的基因编辑的细胞。
[0060]
另一方面,本发明还提供了上述方法制备得到的基因编辑的细胞在制备肿瘤免疫治疗或癌症免疫治疗产品中的应用。
[0061]
在一个实施方式中,上述制备得到的基因编辑的细胞在制备肿瘤免疫治疗或癌症免疫治疗产品时,细胞优选为t细胞;在优选的实施方式中,制备上述基因编辑的t细胞时的供体修复模板包括嵌合抗原受体(car)。
[0062]
进一步的,所述肿瘤或癌症选自黑素瘤、伯基特淋巴瘤、白血病、肉瘤、淋巴瘤、多发性骨髓瘤、脑癌、成神经细胞瘤、成神经管细胞瘤、星形细胞瘤、成胶质细胞瘤、卵巢癌、宫颈癌、子宫癌、结肠直肠癌、乳腺癌、胰腺癌、肺癌、胃癌、甲状腺癌、肝癌、前列腺癌、食道癌、肾癌、膀胱癌和胆囊癌中的一种或任意几种。
[0063]
本发明提供的sgrna能实现pd1基因座的高效率切割,实现外源序列在pd1特定位点的高效整合,有效提高外源序列在pd1特定位点的重组效率,可以增加sgrna在细胞内的稳定性,减少sgrna引起的细胞天然免疫反应,增加细胞存活率。
附图说明
[0064]
此处所说明的图用来提供对本申请的进一步理解,构成本申请的一部分,本申请的示意性实施例及其说明用于解释本申请,并不构成对本申请的不当限定。在图中:
[0065]
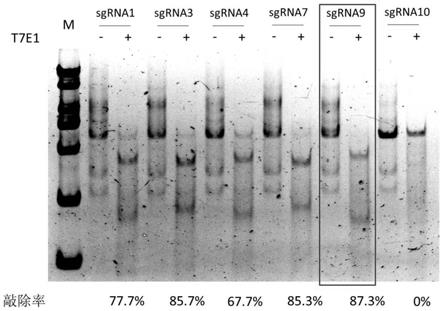
图1 t7e1法筛选pd1高效切割sgrna序列;将靶向pd1的6条不同的sgrna(sgrna1、sgrna3、sgrna4、sgrna7、sgrna9和sgrna10)分别与cas9蛋白混合后导入人t细胞,72h后收集细胞进行t7e1检测。
[0066]
图2流式法筛选pd1高效切割sgrna序列;将靶向pd1的两条sgrna(sgrna7和sgrna9)分别与cas9蛋白混合后导入人t细胞,72h后收集细胞流式检测pd1表达并计算敲除率。
[0067]
图3使用荧光蛋白报告基因作为外源序列,检测定点插入效率;将靶向pd1的两条sgrna(sgrna7和sgrna9)分别与cas9蛋白和荧光蛋白报告基因的外源dna序列混合后导入人t细胞,7天后收集细胞流式检测报告基因整合率。
[0068]
图4在pd1-sgrna9位点可同时实现pd1基因的高效敲除和外源序列的高效整合;将靶向pd1的一条高效sgrna(sgrna9)与cas9蛋白和荧光蛋白报告基因的外源dna序列混合后导入人t细胞,3天后收集细胞流式检测报告基因整合率和pd1敲除率。
[0069]
图5流式检测pd1定点整合cd19-cart阳性率;将靶向pd1的一条高效sgrna(sgrna9)与cas9蛋白和cd19-cart的外源dna序列混合后导入两个不同个体来源的人t细胞,7天后收集细胞流式检测cd19-cart整合率。
[0070]
图6 pd1定点整合cd19-cart的扩增和存活;比较pd1定点整合的cd19-cart细胞与传统慢病毒制备的cd19-cart细胞的体外扩增速率和存活率。
[0071]
图7 pd1定点整合cd19-cart的t细胞激活检测;将pd1定点整合的cd19-cart细胞和慢病毒制备的cd19-cart细胞分别与过表达pdl1的raji肿瘤靶细胞共培养,24h后收集细胞流式检测t细胞激活marker的表达;使用本发明的sgrna可构建pd1敲除的定点整合cd19-cart,与传统慢病毒方法制备的cd19-cart相比,car-t细胞激活程度更好。
[0072]
图8 pd1定点整合cd19-cart的体外杀伤检测;将pd1定点整合的cd19-cart细胞和慢病毒制备的cd19-cart细胞分别与过表达pdl1的raji肿瘤靶细胞共培养,利用ldh法检测体外杀伤;使用本发明的sgrna可构建pd1敲除的定点整合cd19-cart,与传统慢病毒方法制备的cd19-cart相比,car-t细胞具有更好的体外抗肿瘤能力。
具体实施方式
[0073]
结合以下具体实施例和图,对本发明作进一步的详细说明,本发明的保护内容不局限于以下实施例。在不背离发明构思的精神和范围下,本领域技术人员能够想到的变化和优点都被包括在本发明中,并且以所附的权利要求书为保护范围。实施本发明的过程、条件、试剂、实验方法等,除以下专门提及的内容之外,均为本领域的普遍知识和公知常识,本发明没有特别限制内容。如按照sambrook等人,分子克隆,实验室手册(new york:cold spring harbor laboratory press,1989)所记载,或按照厂商的建议条件。
[0074]
1、sgrna的设计:
[0075]
(1)通过ncbi查询人pd1基因组序列,选取1号外显子、2号外显子或关键蛋白编码区进行sgrna设计,所设计的sgrna的靶向序列如下:
[0076]
sgrna1:tgtagcaccgcccagacgac(seq id no.1)
[0077]
sgrna3:gtctgggcggtgctacaact(seq id no.2)
[0078]
sgrna4:aggcgccctggccagtcgtc(seq id no.3)
[0079]
sgrna7:gggcggtgctacaactgggc(seq id no.4)
[0080]
sgrna9:cgactggccagggcgcctgt(seq id no.5)
[0081]
sgrna10:ctacaactgggctggcggcc(seq id no.6)
[0082]
(2)合成各条sgrna的oligo,退火,连接到px458载体。
[0083]
2、靶点的筛选:
[0084]
2-1t7e1酶切:
[0085]
(1)将cas9、sgrna转导入细胞,提取基因组dna;
[0086]
(2)针对sgrna靶点位置设计pcr引物,经过pcr获得包含靶点的dna片段并使用dna切胶回收试剂盒进行纯化;
[0087]
(3)t7e1酶切分析:纯化后的pcr产物进行退火处理,退火程序如下:
[0088]
温度时间95℃5分钟95℃-85℃每秒下降2℃85℃-25℃每秒下降0.1℃
[0089]
退火后产物,加入t7e1酶处理,37℃孵育30分钟,加入dna loading buffer终止反应;
[0090]
(4)聚丙烯酰胺凝胶电泳:将样品加入聚丙烯酰胺凝胶中,电泳槽中加入1xtbe溶液,恒压100v电泳,直至溴酚蓝跑至聚丙烯酰胺凝胶底部。取出凝胶,放入含有eb的tbe溶液中浸泡10分钟,取出聚丙烯酰胺凝胶在紫外光下成像;
[0091]
(5)灰度分析切割率,从中挑选高效率靶点。
[0092]
2-2流式检测敲除率:
[0093]
(1)将cas9、sgrna转导入细胞,2-3天后收集细胞;
[0094]
(2)流式缓冲液清洗细胞一遍后,孵育pd1抗体进行染色,冰上孵育30分钟;
[0095]
(3)用流式缓冲液清洗细胞两遍;
[0096]
(4)用合适体积流式缓冲液重悬细胞,进行流式上机分析;
[0097]
(5)检测pd1表达量的变化,计算敲除率,从中挑选高效率靶点。
[0098]
2-3外源序列重组率检测:
[0099]
(1)将cas9、sgrna以及外源dna转导入细胞,7天后收集细胞;
[0100]
(2)流式缓冲液清洗细胞一遍后,孵育可检测外源蛋白表达的抗体等进行染色,冰上孵育30分钟;
[0101]
(3)用流式缓冲液清洗细胞两遍;
[0102]
(4)用合适体积流式缓冲液重悬细胞,进行流式上机分析;
[0103]
(5)检测外源蛋白表达的细胞比例,从中挑选高重组率靶点。
[0104]
利用针对pd1基因的6条sgrna,sgrna1、sgrna3、sgrna4、sgrna7、sgrna9和sgrna10进行敲除效率的验证,如图1所示,利用t7e1法检测敲除率,sgrna3、sgrna7和sgrna9有较高
敲除率(图1)。
[0105]
继续采用流式的方法,验证sgrna7和sgrna9的敲除率。结果显示两者均有较高敲除率,结果与t7e1一致,如图2所示。
[0106]
之后,将mturquoise2荧光蛋白报告基因作为外源供体序列,检测sgrna7和sgrna9在位点定点插入的效率。结果显示,sgrna7和sgrna9均能起到较高的定点整合效率,其中,sgrna7的定点插入效率达到15.2%,sgrna9的定点插入效率达到23.9%,如图3所示。
[0107]
继续采用利用流式检测sgrna9的定点插入,结果显示,sgrna9位点定点整合的荧光蛋白阳性细胞均为pd1阴性的细胞(如图4所示),进一步证实该方法可实现外源序列的定点整合。
[0108]
3、pd1敲除的增强型cd19-cart细胞的构建:
[0109]
以下以sgrna9为例结合crispr/cas9技术,一步构建pd1敲除的增强型cd19-cart细胞
[0110]
3-1、sgrna9准备:
[0111]
合成sgrna9,溶解于te缓冲液中,稀释成终浓度10ug/ul;
[0112]
3-2、采用电转法制备在pd1定点整合的cd19-cart
[0113]
仪器与材料:
[0114]
①
lonza 4d-nucleofector
tm
system细胞核转仪
[0115]
②
试剂盒为p3primary cell 4d-nucleofector
tm
x kit,lonza,v4xp-3024
[0116]
③
cd3/cd28磁珠刺激后2-3天的t细胞
[0117]
④
商品化spcas9蛋白(10ug/ul)(s.p.cas9nuclease 3nls,idt)
[0118]
⑤
合成的sgrna9
[0119]
具体操作步骤:
[0120]
适用于100μl规格的电转杯:
[0121]
(1)按照82μl solution+18μl supplement/每个电转杯,按电转总数,配置一个电转液mix,混匀,放室温。
[0122]
(2)将cas9蛋白和sgrna9进行共孵育,室温放置10min,形成rnp。
[0123]
(3)rnp中加入“donor”外源供体dna(包含同源臂的外源cd19-cart dna),室温孵育2min。
[0124]
所述cd19-cart包括靶向cd19的胞外结构域,选自cd8α的跨膜区以及选自cd3ζ和cd137的胞内信号传导结构域;另外,在cd19-cart的5
’
和3
’
端设置有同源臂,上下游同源臂序列分别seq id no.7和seq id no.8所示。
[0125]
(4)收集激活状态的t细胞,计数为5
×
10
6
进行一个电转反应。
[0126]
(5)将细胞与“rnp+donor”充分混合重悬,加入电转杯。
[0127]
(6)打开电转仪,将电转杯放入槽孔,选择相应程序(stimulated human t cell)进行电转。
[0128]
(7)将细胞加入已预热的细胞培养基中,于细胞培养箱中培养。
[0129]
3-3、对pd1定点整合cd19-cart细胞进行评价
[0130]
将上述“donor”外源供体dna作为外源dna序列,在两个不同供体(donor-1和donor-2)的t细胞中证实了其在pd1-sgrna9位点均具有较高的定点整合率,如图5所示,定
点整合效率达到20%-30%。
[0131]
另外,采用pd1定点整合cd19-cart的细胞(pd1-cd19-cart)与传统慢病毒制备的cd19-cart细胞(cd19-cart(lenti))相比,细胞扩增速率相当;但是,采用pd1定点整合cd19-cart的细胞与传统慢病毒制备的cd19-cart细胞相比,表现出了更高的细胞存活率,如图6所示。
[0132]
与过表达pdl1的raji肿瘤细胞(伯基特淋巴瘤细胞)共培养,通过流式检测t细胞激活标志物cd69和cd137表达,结果显示pd1定点整合cd19-cart细胞(pd1-cd19-cart)与传统慢病毒制备的cd19-cart细胞(cd19-cart(lenti))相比,具有相似的cd69表达以及更高的cd137表达,如图7所示。
[0133]
利用ldh法检测体外杀伤,观察到pd1定点整合的cd19-cart细胞(pd1-cd19-cart)与传统慢病毒制备的cd19-cart细胞(cd19-cart(lenti))相比,具有更强的杀伤过表达pdl1的raji肿瘤细胞能力,如图8所示。
[0134]
综上,使用本发明的sgrna,结合crispr/cas9技术,可一步构建pd1敲除的定点整合cd19-cart细胞。该方法与传统慢病毒方法相比,可减少car-t制备过程中使用病毒带来的高昂成本,极大降低car-t疗法的治疗费用。另一方面,该方法使car-t元件定向插入pd1基因座的特定位点,可减少病毒随机插入带来的安全隐患。此外,此方法可一步构建pd1敲除的增强型cd19-cart细胞,能提高car-t细胞的抗肿瘤能力。该实施例证明了本发明保护的sgrna的重要性和价值,但不限于cd19-cart外源序列在pd1特定位点的定向插入,可拓展到其他cart序列以及用于免疫治疗其他疗法的开发。
[0135]
以上所述仅为本申请的实施例而已,并不用于限制本申请。对于本领域技术人员来说,本申请可以有各种更改和变化。凡在本申请的精神和原理之内所作的任何修改、等同替换、改进等,均应包含在本申请的权利要求范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1