一种瑞卢戈利药物中间体的制备方法与流程
[0001]
本申请涉及药物合成领域,具体地,涉及一种表现出促性腺素释放激素(gnrh)拮抗活性的噻吩并[2,3-d]嘧啶化合物及其中间体的制备方法。
背景技术:
[0002]
子宫内膜异位症是子宫内膜生长在子宫腔以外的任何部位所引起的,是一种常见的雌激素依赖的妇科疾病,常发生于女性生育年龄期间,其作用机制尚不清楚。子宫内膜异位症的诊断困难及病因不明等复杂症状,严重阻滞了其有效治疗方法的发现。目前,子宫内膜异位症主要通过腹腔镜手术诊断,并通过外科手术进行治疗,或服用避孕药,gnrh受体激动剂或孕激素减少体内雌激素水平来进行控制。2018年7月该领域全球首个口服gnrh拮抗剂依拉戈利钠获得fda批准上市。
[0003]
relugolix是由日本武田药品株式会社开发研制的小分子促性腺激素释放素(gnrh)受体拮抗剂,能够迅速降低女性雌激素和孕激素。2019年1月relugolix在日本获得批准上市,被批准用于子宫肌瘤的治疗和症状缓解。预计2019年第三季度会在fda递交新药申请。
[0004]
目前为止,关于relugolix的合成工艺国内外相关报道较少,原研公司武田药品株式会社首次披露了其合成路线(wo2004067535a1)。具体合成路线如下所示:专利wo2014051164a2公布的relugolix的另一种合成路线如下:
上述两条披露的合成路线中合成过程用到了剧毒性物质氯甲酸乙酯,且闪点很低,高度易燃,对于生产环境要求较高。
技术实现要素:
[0005]
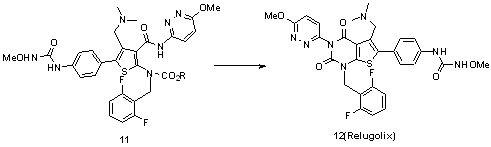
本申请,提供一种新的瑞卢戈利的合成方案,其合成路线如下:首先,化合物1和氯甲酸酯反应得到化合物2。化合物2和化合物3发生亲核取代反应得到化合物4。化合物4经过溴化反应得到化合物5。化合物5与盐酸二甲胺发生取代反应生成化合物6。化合物6在碱性条件下水解得到化合物7。化合物7和化合物8发生缩合反应生成化合物9。化合9发生还原反应得到化合物10。化合物10与盐酸甲氧基胺发生缩合反应生成化
合物11。化合物11发生分子内关环反应得到化合物12(relugolix)。
[0006]
与现有技术相比,本申请用于合成瑞卢戈利中间体的方法具有以下益处:(1)避免了高毒、低闪点、高度易燃物质氯甲酸乙酯和氯甲酸甲酯的使用;(2)可以有效的避免氯甲酸乙酯和氯甲酸甲酯存储及使用过程存在的风险。
[0007]
(3)与wo2014051164a2路线相比,将pd/c催化氢化步骤前移,避免了重金属对最终产品污染的风险,也更有利于cgmp条件下原料药的生产。
[0008]
(4)与wo2004067535a1路线相比,更加简洁高效,避免了甲氧乙基甲基胺的取代反应,更加安全环保。
[0009]
具体的实施方式下面通过实施例来描述本申请的实施方式,本领域的技术人员应当认识到,这些具体的实施例仅表明为了达到本申请的目的而选择的实施技术方案,并不是对技术方案的限制。根据本申请的教导,结合现有技术对本申请技术方案的改进是显然的,均属于本申请保护的范围。
[0010]
实施例中采用的实施条件可以根据具体要求做进一步调整,未注明的实施条件通常为常规实验中的条件。其中,在以下实施例中用到的化学试剂均为市购的化学试剂。
[0011]
实施例12 l反应瓶中依次加入甲苯(450 ml),化合物1(150 g),开启搅拌,逐滴加入氯甲酸丙酯(120 g), 加热回流反应2 h。降温至50~60
ꢀ°
c,控制内温在50~60
ꢀ°
c滴加乙醇(1350 ml)。滴加完毕,缓慢降温至内温0~10
ꢀ°
c,搅拌1 h,过滤,滤饼用乙醇(300 ml)淋洗。45
ꢀ°
c真空干燥,得到黄色固体,收率84.4%,纯度98 %。
[0012]
化合物2-a核磁数据如下:1h nmr (400 mhz, cdcl3) δ 10.67 (s, 1h), 8.31-8.22 (m, 2h), 7.59-7.52 (m, 2h), 4.39 (q, j = 7.1 hz, 2h), 4.21 (t, j = 6.7 hz, 2h), 2.42 (s, 3h), 1.80-1.67 (m, 2h), 1.42 (t, j = 7.1 hz, 3h), 1.00 (t, j = 7.4 hz, 3h)。
[0013]
实施例210 ml反应瓶中依次加入甲苯(1.5 ml),化合物1(0.5 g),开启搅拌,逐滴加入氯甲酸异丙酯(0.4 g), 加热回流反应2 h。降温至20~30
ꢀ°
c,加入纯净水(4 ml)和二氯甲烷(4 ml),分层分液后水相用二氯甲烷(4 ml)萃取,合并有机相,减压浓缩得到化合物2-b粗品,过柱纯化(乙酸乙酯:正庚烷=1:10),得到亮黄色固体0.58 g,产率90%,纯度98%。
7.1 hz, 3h), 1.07-0.71 (m, 3h)。
[0019]
实施例55 l反应瓶中依次加入乙酸乙酯(1.64 l)、三氟甲基苯(1.64 l),开启搅拌,加入化合物4-a(211g),加入n-溴代丁二酰亚胺(90.7g),加入2,2-偶氮二(2,4-二甲基戊腈)(10.1 g),外温65~75
ꢀ°
c下搅拌加热反应1 h。反应完毕,降至室温,加入乙酸乙酯(633 ml),用水(844 ml)洗涤两次,收集有机相,减压浓缩至750~800 ml,加入乙醇(2 x 576 ml)套蒸两次。向体系中加入正庚烷(1.6 vol),上述混合物在20~30
ꢀ°
c下搅拌30 min,再次加入正庚烷(1.2 vol),降温至0~10
ꢀ°
c搅拌1 h,过滤,滤饼用乙醇和正庚烷混合溶液(1:2,337 ml)洗涤。滤饼45
ꢀ°
c真空干燥,得到浅黄色固体,收率73%,hplc纯度98.7%。
[0020]
化合物5-a核磁数据如下:1h nmr (400 mhz, cdcl3) δ 8.43-8.27 (m, 2h), 7.81-7.65 (m, 2h), 7.36-7.18 (m, 1h), 6.87 (t, j = 7.8 hz, 2h), 4.99 (s, 2h), 4.71 (s, 2h), 4.32 (q, j = 6.9 hz, 2h), 4.08 (br, 2h), 1.61 (br, 3h), 1.37 (t, j = 7.2 hz, 3h), 1.10-0.71 (m, 3h)。
[0021]
实施例6100 ml反应瓶中依次加入乙酸乙酯(4 ml)、三氟甲基苯(4 ml),开启搅拌,加入化合物4-a(0.5 g),加入液溴(0.193 g),加入2,2-偶氮二(2,4-二甲基戊腈)(0.024 g),外温65~75
ꢀ°
c下搅拌加热反应22 h, 反应液产品峰面积34.7%。
[0022]
实施例7
100 l反应瓶中依次加入乙酸乙酯(4 ml)、三氟甲基苯(4 ml),开启搅拌,加入化合物4-a(0.5 g),加入二溴海因(0.172 g),加入2,2-偶氮二(2,4-二甲基戊腈)(0.024 g),外温65~75
ꢀ°
c下搅拌加热反应1 h, 反应液产品峰面积91%。
[0023]
实施例83 l反应瓶中依次加入n,n-二甲基甲酰胺(962 ml),盐酸二甲胺(35.8 g),三乙胺(77.1 g),开启搅拌,上述混合物在20~30
ꢀ°
c搅拌0.5 h。内温降至0~10
ꢀ°
c,加入化合物5-a(175 g)和n,n-二甲基甲酰胺(87.5 ml),在10~20
ꢀ°
c下搅拌1 h。反应完毕,加入乙酸乙酯(875 ml),水(875 ml),搅拌,分液,收集有机相。水相用乙酸乙酯(525 ml)萃取一次,合并有机相。有机相用10% 食盐水(3 x 875 ml)洗涤,用水(875 ml)洗涤一次,有机相减压浓缩至无液体蒸出,得到化合物6-a粗品, hplc纯度92%,直接用于下一步反应。取部分过柱纯化用于核磁数据表征。
[0024]
化合物6-a核磁数据如下:1h nmr (400 mhz, cdcl3) δ 8.31-8.18 (m, 2h), 7.70-7.61 (m, 2h), 7.32-7.19 (m, 1h), 6.92-6.78 (m, 2h), 5.02 (s, 2h), 4.23 (q, j = 7.2 hz, 3h), 4.19-4.05 (m, 2h), 3.52 (s, 2h), 2.06 (s, 6h), 1.77-1.50 (m, 3h), 1.32 (t, j = 7.1 hz, 3h), 0.88 (t, j = 6.8 hz, 3h)。
[0025]
实施例9
5 l反应瓶中依次加入乙醇(1.5 l),水(450 ml),化合物6-a(173 g),加入48%的氢氧化钾水溶液(54 g),加热至55~65
ꢀ°
c反应5 h。降温至20~30
ꢀ°
c,用6 mol/l 盐酸调ph至6.0~7.0,减压浓缩混合液体积小于605 ml,加入二氯甲烷(865 ml),然后加入水(865 ml);搅拌,分液,收集下层有机相,水相用二氯甲烷(2 x 519 ml)萃取,合并有机相。有机相用10% 食盐水(865 ml)洗涤一次,用水(865 ml)洗涤一次,减压浓缩至约175 ml。乙酸乙酯(2 x 692 ml)套蒸至175 ml,补加346ml乙酸乙酯。20~30
ꢀ°
c,搅拌3 h,过滤,滤饼用冷的乙酸乙酯(346 ml)淋洗,滤饼在40~50
ꢀ°
c下干燥 ,得到黄色固体,收率86%,hplc纯度94.3%。
[0026]
化合物7-a核磁数据如下:1h nmr (400 mhz, cdcl3) δ 8.29 (d, j = 8.5 hz, 2h), 7.44 (d, j = 8.6 hz, 2h), 7.33-7.15 (m, 1h), 6.94-6.74 (m, 2h), 5.08 (s, 2h), 4.26-3.98 (m, 2h), 3.79 (s, 2h), 2.41 (s, 6h), 1.83-1.49 (m, 2h), 1.11-0.66 (m, 3h)。
[0027]
实施例102 l反应瓶中依次加入n,n-二甲基乙酰胺(550 ml),化合物7-a(110 g)和化合物8(39.98 g),n2保护,控制温度在10~40
ꢀ°
c滴入n,n-二异丙基乙胺(66.6 g),升温到50~60
ꢀ°
c反应0.5 h。控制温度不高于60
ꢀ°
c滴加50% t3p的乙酸乙酯溶液(157.4 g),加毕在50~60
ꢀ°
c下搅拌反应1 h。降温,控制内温20~30
ꢀ°
c,滴入水(825 g)。控制内温20~30
ꢀ°
c用8 mol/l氢氧化钠水溶液调ph至7.5~8.5,搅拌0.5 h,过滤,滤饼加入到甲醇(440 ml)中,在20~30
ꢀ°
c搅拌2 h以上,过滤,滤饼用甲醇(220 ml)淋洗。45
ꢀ°
c真空烘料,得黄色固体,收率78.1%,纯度99.3%。
[0028]
化合物9-a核磁数据如下:1h nmr (400 mhz, cdcl3) δ 13.87 (s, 1h), 8.55 (d, j = 9.5 hz, 1h), 8.29 (d, j = 8.6 hz, 2h), 7.567.45 (m, 2h), 7.207.07 (m, 1h), 7.00 (d, j = 9.6 hz, 1h), 6.75 (t, j = 7.7 hz, 2h), 5.04 (s, 2h), 4.37-3.99 (m, 5h), 3.51 (s, 2h), 2.20 (s, 6h), 1.85-1.49 (m, 2h), 1.09-0.85 (m, 3h).化合物9-a的质谱数据:[m+h]
+
=641.3。
[0029]
实施例11
50 ml反应瓶中依次加入甲醇(16 ml),化合物9-a(2 g),浓盐酸(0.27 g),氢气压力0.1~0.3 mpa,控制温度20~30
ꢀ°
c反应15 h。反应结束,用硅藻土过滤,滤饼用甲醇(4 ml)淋洗,母液减压浓缩至无馏分蒸出,加入二氯甲烷(20 ml)和饱和碳酸氢钠溶液(10 ml),分层分液后水相用二氯甲烷(20 ml)萃取,合并有机相,减压浓缩得到化合物10-a粗品,过柱纯化(乙酸乙酯:正庚烷=3:2),得到亮黄色固体1.62 g,产率85%,纯度97%。
[0030]
化合物10-a核磁数据如下:1h nmr (400 mhz, dmso-d6) δ 14.05 (s, 1h), 8.38 (d, j = 9.5 hz, 1h), 7.38-7.23 (m, 2h), 7.04-6.89 (m, 4h), 6.67-6.57 (m, 2h), 5.41 (s, 2h), 4.89 (s, 2h), 4.00 (s, 5h), 3.54 (s, j = 21.9 hz, 2h), 2.10 (s, 6h), 1.71-1.37 (d, 2h), 0.73 (d, 3h).化合物10-a的质谱数据:[m+h]
+
=611.2。
[0031]
实施例1210 ml反应瓶中依次加入乙腈(2 ml),三乙胺(28.1 mg),n,n-羰基二咪唑(102 mg),氮气置换三次,体系冰水浴下加入盐酸甲氧基胺(53 mg),氮气置换三次,加入化合物10-a(198 mg),氮气置换三次,体系45~55
ꢀ°
c反应2 h,加入三乙胺(42 mg)和n,n-羰基二咪唑(102 mg)继续反应15 h,反应结束,加入二氯甲烷(2 ml)和纯净水(2 ml),分层分液后水相用二氯甲烷(2 ml)萃取,合并有机相,浓缩,过柱纯化(乙酸乙酯:正庚烷=2:1),得到类白色固体118 mg,产率52%,纯度87%。
[0032]
化合物11-a核磁数据如下:1h nmr (400 mhz, dmso-d6) δ 14.00 (s, 1h), 9.62 (s, 1h), 9.07 (s, 1h), 8.38 (d, j = 9.5 hz, 1h), 7.83-7.62 (m, 2h), 7.43-7.11 (m, 4h), 6.98 (t, j = 7.9 hz, 2h), 4.91 (s, 2h), 4.00 (s, 5h), 3.64 (s, 3h), 3.54 (d, j = 3.1 hz, 3h), 2.10 (s, 6h), 1.43 (d, j = 18.2 hz, 2h), 0.79 (d, j = 42.4 hz, 3h).化合物11-a的质谱数据:[m+h]
+
=684.1。
[0033]
实施例1310 ml反应瓶中依次加入化合物11-a(100 mg),甲醇(2.5 ml),四氢呋喃(0.2 ml),甲醇钠(4 mg),体系氮气置换三次,控制温度60~65
ꢀ°
c反应2 h,反应结束,加入二氯甲烷(2 ml)和纯净水(2 ml),分层分液后水相用二氯甲烷(2 ml)萃取,合并有机相,减压浓缩,过柱纯化(乙酸乙酯:甲醇=40:1),得到类白色固体19.7 mg,产率21.6%,纯度95%。
[0034]
化合物12核磁数据如下:1h nmr (400 mhz, dmso-d6) δ 9.66 (s, 1h), 9.10 (s, 1h), 7.75 (dd, j = 12.4, 8.9 hz, 3h), 7.50 (dd, j = 27.9, 8.8 hz, 4h), 7.15 (t, j = 8.1 hz, 2h), 5.31 (dd, j = 63.3 hz, 2h), 4.10 (s, 3h), 3.65 (s, 5h), 2.05 (s, 6h).化合物12的质谱数据:[m+h]
+
=624.1。
[0035]
本申请包括但不限于以上实施例,凡是在本申请精神的原则下进行的任何等同替代或局部改进,都将视为在本申请的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1