基于tRNA-gRNA-cRNA进行日本青鳉胚胎和细胞的基因编辑系统的制作方法
基于trna-grna-crna进行日本青鳉胚胎和细胞的基因编辑系统
技术领域
[0001]
本发明属于分子生物学领域,具体涉及一种方法,即利用rna聚合酶启动子cmv在体内驱动trna-sgrna系统稳定表达sgrna,结合cas9表达载体或者cas9蛋白可以在鱼类细胞和胚胎内进行基因编辑。
背景技术:
[0002]
成簇规律间隔的短回文序列(clustered regularly interspaced short palindromic repeat,crispr)和crispr相关蛋白(crispr-associated protein,cas)形成的crispr/cas系统是古生菌体内一种重要的获得性免疫系统(jinek et al.,2012)。作为该系统的成员之一,经优化的crispr/cas9系统已经发展成为基因编辑技术的有力工具。crispr/cas9系统在实现基因编辑的过程中利用cas9蛋白在sgrna的引导下对基因组dna进行切割,产生非同源末端连接和同源末端修复(mali et al.,2013;ran et al.,2013),从而在真核生物、原核生物以及病毒基因组中实现靶点位置附近序列的突变(jiang et al.,2013;platt et al.,2014;tang et al.,2017;wefers et al.,2017;a.xu et al.,2015。
[0003]
为了利用crispr/cas9系统在生物体内实现多基因的敲除或者在特定组织中的基因编辑,大多数研究者通常是利用rna聚合酶iii类启动子(如:u6,7sk,h1)驱动sgrna的表达实现多基因编辑,或者利用组织特异的rna聚合酶ii类启动子驱动cas9蛋白的特异表达实现在特定组织中的基因编辑(chen et al.,2017;merenda et al.,2017)。然而相关的研究表明这些启动子的序列或者功能在物种间并不保守。例如在日本青鳉中,利用人类的u6启动子不能驱动sgrna的表达(liu et al.,2018)。近来,新的策略被运用于在体内产生具有功能的sgrna。第一种策略是利用核酸内切酶csy4的靶序列连接的sgrna序列能被csy4蛋白切割形成功能性的sgrna,这一策略已经在酵母,高等植物,斑马鱼以及人类细胞系中成功实现基因敲除(cermak et al.,2017;ferreira et al.,2018;nissim et al.,2014;qin et al.,2015)。然而,共注射了csy4 mrna,cas9 mrna和csy4-grna的斑马鱼胚胎展现出严重的畸形,这说明了csy4蛋白对斑马鱼胚胎是有严重的毒性((qin et al.,2015)。第二种策略是利用核酶(ribozyme)对核酶连接的sgrna序列进行切割形成sgrna,并且在rna聚合酶ii类启动子的调控下诱导条件性基因敲除(he et al.,2017)。在这种策略中,由于锤头型核酶(hammerhead ribozyme,hh)具有5’末端切割活性而丁型肝炎病毒核酶(hepatitis delta virus ribozyme,hdv)具有3’末端切割活性,因而需要被同时运用到该方法中去切割含有核酶和sgrna序列的转录本(avis et al.,2012)。这两种核酶的序列较长且不相同,该方法在构建表达质粒时也因此变得较为复杂。最新且最便捷的策略是利用trna序列的5’端和3’端能被rnase p和rnase z分别切割的特性(forster&altman,1990;schiffer et al.,2002),因此trna连接多个sgrna的序列长度比核酶连接sgrna的序列长度更短,此方法已经在植物、果蝇、斑马鱼以及人类细胞中实现了多个基因的敲除(knapp et al.,2019;port&bullock,2016;qi et al.,2016;shiraki&kawakami,2018;xie et al.,2015)。此外,
在组织特异启动子的调控下,该方法在小鼠体内实现特定组织的基因敲除(xu et al.,2017)。毫无疑问,trna-grna系统是目前介导多基因敲除或组织条件性敲除的最佳解决方案。
[0004]
近来,crispr/cas9系统已经被运用在日本青鳉中获得突变体,主要通过共注射sgrna和优化的cas9 mrna到1细胞期的胚胎中(fang et al.,2018)。然而,多基因敲除或组织条件性敲除的方法在日本青鳉中还没有建立,而且能在鱼类细胞中表达有活性的grnas还是一个难题。
技术实现要素:
[0005]
本发明的目的在于提供了一种在青鳉胚胎或细胞中表达grna的序列单元;所述的单元是trna(seq id no.1所示序列)、目的基因grna和crna(seq id no.2所示序列)依次连接而得。
[0006]
本发明的另一个目的在于提供了上述序列单元的应用,将上述的n个序列单元串联(n为1或2或3)插入鱼类表达质粒载体后,可在青鳉内产生功能性的grna,因此可用于青鳉的基因编辑。
[0007]
本发明的最后一个目的在于提供了一种青鳉胚胎或细胞的基因编辑方法,本发明的方法简单,可同时对多个基因进行基因编辑,编辑效率高。
[0008]
为了达到上述目的,本发明采取以下技术措施:
[0009]
一种在青鳉胚胎或细胞中表达grna的序列单元;所述的单元是trna(seq id no.1所示序列)、目的基因grna和crna(seq id no.2所示序列)依次连接而得。
[0010]
一种在青鳉胚胎或细胞中表达grna的序列单元的应用,所述的序列单元可用于青鳉胚胎或精原干细胞的基因编辑;
[0011]
以上所述的应用中,优选的,是将上述的n个序列单元串联(n为1或2或3)后插入鱼类表达质粒载体,得到的载体与线性化的cas9 mrna显微注射入青鳉胚胎或与表达cas9蛋白的质粒转染精原干细胞,实现目的基因的编辑。
[0012]
一种青鳉胚胎或细胞的单基因编辑方法,包括下述步骤:
[0013]
1)trna(seq id no.1所示序列)、目的基因grna和crna(seq id no.2所示序列)依次连接,获得trna-grna-crna;
[0014]
2)trna-grna-crna插入到经bamhi+xho i酶切的pcs2+质粒中,构成pcs2-trna-grna质粒;
[0015]
3)pzcas9质粒进行线性化,体外转录合成zcas9 mrna,与trna-grna-crna一起显微注射进入1细胞期的青鳉胚胎中;或pzcas9质粒与pcs2-trna-grna质粒混合后转染青鳉精原干细胞。
[0016]
以上所述的方法中,优选的,当目的基因为tyr时,trna-grna-crna的序列为:seq id no.7或seq id no.3所示。
[0017]
一种青鳉胚胎或细胞的双基因编辑方法,是将单基因编辑方法中的步骤1)替换成:trna-grna1-crna和trna-grna2-crna串联,得到grna1-crna-trna-grna2;grna1和grna2代指其他的不同的目标基因的grna;其余操作相同。
[0018]
与现有技术相比,本发明具有以下优点:
[0019]
1、本发明利用rna聚合酶启动子cmv稳定表达sgrna。
[0020]
2、trna对鱼类细胞和胚胎基本没有毒性,并能产生有功能性的grna。
[0021]
3、构建的pcs2-trna-tyr grna可以在体外转录grna,或者在体内稳定表达grna,而且在该质粒中间的克隆位点(bbsi-bbsi)方便外源片段grna的插入,这样设计的bbsi-bbsi位点可以去除trna-crna中间的所有碱基,不会残留碱基,而可以用来直接克隆其他的基因靶位。
[0022]
4、构建的载体可以产生crna-trna融合片段,为双基因的敲除提供了模板,可以设计任何双基因(grna1/grna2,grna1和grna2代指其他的不同的目标基因的grna)的靶位点进行简单的pcr扩增而获得双基因的敲除片段grna1-crna-trna-grna2并进行敲除质粒的构建,通过简单的一步融合infusion-pcr就可以克隆到pcs2-trna-tyr grna(cmv-sp6-trna-mcs-crna-trna-tyrgrna-crna)中间,获得含有多个grna的载体(cmv-sp6-trna-grna1-crna-trna-grna2-crna-trna-tyrgrna-crna)从而对双基因或者三基因进行敲除。
附图说明
[0023]
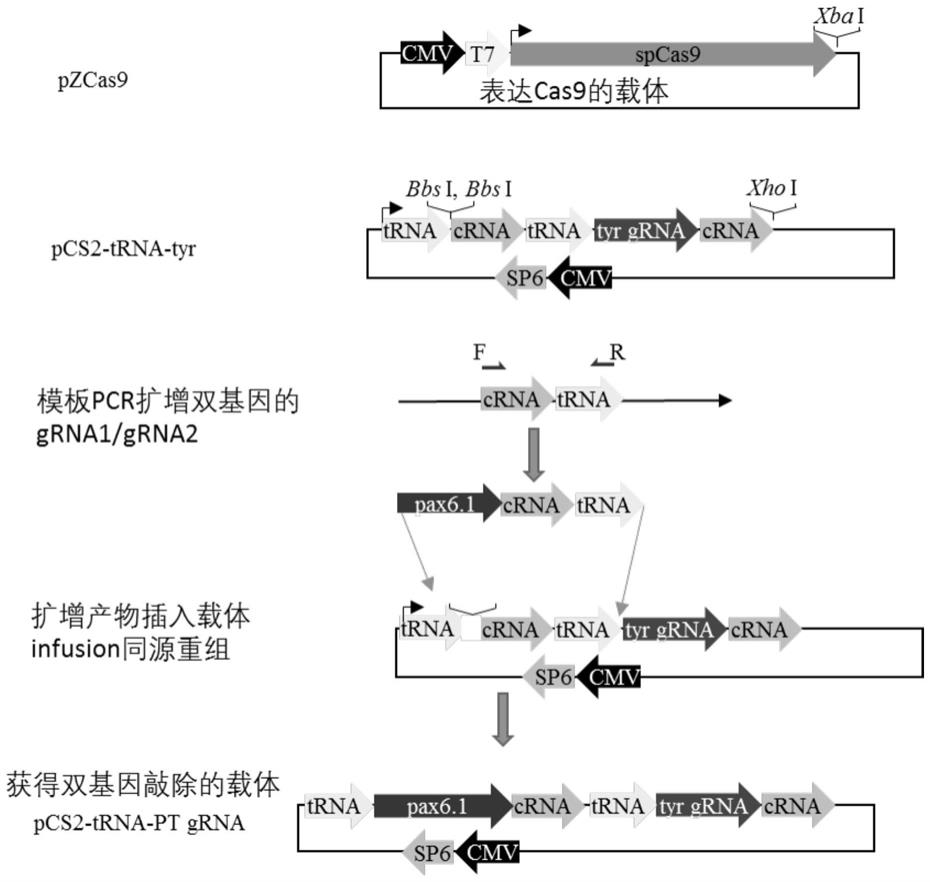
图1为青鳉中单基因(tyr)和双基因(pax6.1,tyr)编辑载体的构建示意图。
[0024]
图2为青鳉胚胎和sg3细胞中对tyr基因进行编辑后获得的突变体的序列及测序结果。
[0025]
图3为对胚胎中的tyr基因编辑后获得可以繁殖的突变体。
[0026]
图4为双编辑了pax6.1和tyr基因后青鳉胚胎的观察和序列分析;
[0027]
图中显示,青鳉胚胎从表型上看有突变,通过测序进一步从dna序列上确认。
具体实施方式
[0028]
本发明所述技术方案,如未特别说明,均为本领域的常规方式;所用试剂或材料,如未特别说明,均来源于商业渠道。本发明以日本青鳉的tyr为例进行说明,利用其它基因pax6.1和tyr也能完成编辑目的。
[0029]
实施例1:
[0030]
基于trna序列进行日本青鳉胚胎的单基因编辑:
[0031]
1.实验材料
[0032]
野生型orange-red品系日本青鳉饲养于华中农业大学水产学院实验教学基地28℃恒温水循环系统中,光照周期为14h光照,10h无光处理。显微注射所用胚胎由雌雄青鳉自然产卵所得。青鳉精原干细胞sg3(hong et al.,2004)由本实验室保存并在28℃培养箱中进行培养。
[0033]
2.质粒构建
[0034]
将trna-mcs-crna-trna-tyr grna-crna(seq id no.3)序列送天一辉远生物科技有限公司(http://www.dna1953.com.cn/index.html)人工合成,随后将该片段插入到经bamh i+xho i酶切的pcs2+质粒(yu et al.,2017)中,构成pcs2-trna-tyr grna质粒(图1,seq id no.4)。本步骤使用seq id no.3所示序列是为了构成pcs2-trna-tyr grna质粒以作为实施例2中,引入新的靶基因grna的模板。
[0035]
3.显微注射
[0036]
利用xbai对pzcas9质粒(fang et al.,2018)进行线性化,随后用mmessagemmac hine
tm
t7 transcription kit试剂盒进行体外转录合成zcas9 mrna。将终浓度为500ng/μl的zcas9 mrna、30ng/μl的pcs2-trna-tyr grna质粒和0.05%的酚红混合溶液用picolitermicroinjector注射仪(warner,美国)注射到1细胞期的胚胎中。注射过的胚胎用erm培养基置于28℃恒温培养箱中培养(chen et al.,2017)(fang et al.,2018)。注射第五天时用leica m205 fa显微镜对眼部色素缺失情况进行观察及拍照。
[0037]
4.细胞培养与转染
[0038]
将生长良好的青鳉精原干细胞sg3均匀铺在12孔板中,细胞密度为5
×
105,待细胞生长至70%时进行转染。转染时将细胞培养基吸出,加入1ml opti-mem培养基;取两个无酶无菌ep管,一个加入50μlopti-mem和1μg质粒(pcs2-trna-tyr grna:pzcas9=1:1),另外一个加入50μlopti-mem和3μl的lipofectamine 2000,室温孵育5min;将上述两个ep管中的混合液合并,室温孵育10min;将合并的混合液加入到细胞孔中,轻轻晃动细胞平板混匀,放入培养箱中3h后将opti-mem培养基换成普通培养基正常培养(xue et al.,2018)。每隔2天转染一次,共转染3次。转染完成后,置于倒置荧光显微镜下观察并拍照。
[0039]
5.突变检测
[0040]
收集表型突变的胚胎、野生型胚胎、转染的细胞和未转染的细胞分别提取基因组dna。提取胚胎基因组dna时只需单个胚胎即可。将收集的胚胎或者细胞放入1.5ml离心管中,分别加入600ul细胞裂解液和10μl 50ng/μl的蛋白酶k,65℃孵育4h,待胚胎或则细胞完全裂解之后取出室温冷却,加入200μl7.5m的醋酸铵上下颠倒混匀置于冰上5min,12000rpm 4℃离心10min;取上清液600μl至新管,加入600μl异丙醇,轻摇析出沉淀物,静置1-2min;12000rpm 4℃离心10min,弃上清液;加入75%乙醇1ml轻摇混匀,12000rpm 4℃离心5min,弃上清液;加入100%乙醇1ml,12000rpm 4℃离心5min,弃上清液,空气中干燥沉淀呈半透明状时加入适量的te溶液进行溶解,测浓度,-20℃保存(wang et al.,2011)。
[0041]
对tyr grna靶位点附近的片段进行扩增。引物为tyr-f:5’cgagtacgcctacctgtt和tyr-r:5’ctagatgtggtcggtgaga。扩增体系如下1μl 100ng/μl的gdna,1μl10μm的tyr-f+tyr-r,10μltaq酶mix,无菌水8μl。扩增条件为94℃3min;94℃30s,60℃30s,72℃30s,30个循环;72℃5min,4℃保存。琼脂凝胶电泳检测后,切胶用胶回收试剂盒纯化pcr扩增产物。将回收的dna片段连接pmd18-t载体,连接产物转化到dh5α感受态细胞中,涂板。挑取10个克隆进行菌液pcr鉴定,之后送天一辉远生物科技有限公司测序。测序结果用snapgene软件(https://www.snapgene.com/)进行分析。通过序列分析,共有5个克隆显示序列突变,基因突变主要发生在靶序列附近,从而导致多种移码突变;
[0042]
胚胎和sg3细胞的突变和测序的部分结果,具体如图2所示;获得的突变胚胎显示白化性状,且可繁殖(图3)。
[0043]
上述实施方案,将trna-mcs-crna-trna-tyr grna-crna(seq id no.3)替换为trna-tyr grna-crna(seq id no.7所示序列)也能完成基因编辑。
[0044]
实施例2:
[0045]
基于trna序列进行日本青鳉胚胎的双基因(pax6.1和tyr)编辑:
[0046]
1.bbsi酶切质粒pcs2-trna-tyr grna后,以此为模板,利用引物f:5’ttcccggctggtgcatgcctggtggaatccggcaggttttagagctagaaatagc和r:5’ttctagctctaaaactcatctgtgg
ctctggactgtgcaccagccgggaatcgaa进行扩增(基因pax6.1的grna通过引物引入),获得seq id no.5所示序列,利用ii one step cloning kit(c112-01,vazyme)将扩增片段插入经bbsi酶切后的pcs2-trna-tyr grna质粒中,从而获得双基因编辑的载体pcs2-trna-pt grna(seq id no.6)。
[0047]
2.显微注射
[0048]
利用xbai对pzcas9(fang et al.,2018)质粒进行线性化,随后用mmessagemmac hine
tm
t7 transcription kit试剂盒进行体外转录合成zcas9 mrna。将终浓度为500ng/μl的zcas9 mrna、30ng/μl的pcs2-trna-pt grna质粒和0.05%的酚红混合溶液用picolitermicroinjector注射仪(warner,美国)注射到1细胞期的胚胎中。注射过的胚胎用erm培养基置于28℃恒温培养箱中培养(chen et al.,2017)(fang et al.,2018)。注射第五天时用leica m205 fa显微镜对眼部色素缺失情况进行观察及拍照。
[0049]
3.突变检测
[0050]
收集表型突变的胚胎、野生型胚胎分别提取基因组dna。提取胚胎基因组dna时只需单个胚胎即可。将收集的胚胎放入1.5ml离心管中,分别加入600ul细胞裂解液和10μl50ng/μl的蛋白酶k,65℃孵育4h,待胚胎或则细胞完全裂解之后取出室温冷却,加入200μl7.5m的醋酸铵上下颠倒混匀置于冰上5min,12000rpm 4℃离心10min;取上清液600μl至新管,加入600μl异丙醇,轻摇析出沉淀物,静置1-2min;12000rpm 4℃离心10min,弃上清液;加入75%乙醇1ml轻摇混匀,12000rpm 4℃离心5min,弃上清液;加入100%乙醇1ml,12000rpm 4℃离心5min,弃上清液,空气中干燥沉淀呈半透明状时加入适量的te溶液进行溶解,测浓度,-20℃保存(wang et al.,2011)。
[0051]
对tyr grna靶位点附近的片段进行扩增。引物为tyr-f:5’cgagtacgcctacctgtt和tyr-r:5’ctagatgtggtcggtgaga;
[0052]
对pax6.1靶位点附近的片段进行扩增,引物为pax6.1-f:5’tttgctcattgatactgttgtgggt和pax6.1-r:5’atgtcatgcctttgttgccc;
[0053]
扩增体系如下1μl 100ng/μl的gdna,1μl 10μm的tyr-f+tyr-r,10μltaq酶mix,无菌水8μl。扩增条件为94℃3min;94℃30s,60℃30s,72℃30s,30个循环;72℃5min,4℃保存。琼脂凝胶电泳检测后,切胶用胶回收试剂盒纯化pcr扩增产物。将回收的dna片段连接pmd18-t载体,连接产物转化到dh5α感受态细胞中,涂板。挑取单克隆进行菌液pcr鉴定,之后送天一辉远生物科技有限公司测序。测序结果用snapgene软件(https://www.snapgene.com/)进行分析。通过序列分析,tyr基因挑取的8个克隆有1个发生突变,pax6.1基因挑取48个克隆有3个发生突变,突变主要发生在靶序列附近,从而导致多种移码突变。
[0054]
双基因编辑后的突变胚胎表型如图4所示。双基因敲除中所挑选的胚胎进行检测,结果是双基因突变的。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1