化合物、其应用及包含其的有机电致发光器件的制作方法
[0001]
本发明涉及有机发光材料技术领域,具体而言,涉及一种化合物、其应用及包含其的有机电致发光器件。
背景技术:
[0002]
有机电致发光(oled:organic light emission diodes)器件是一类具有类三明治结构的器件,包括正负电极膜层及夹在电极膜层之间的有机功能材料层。对oled器件的电极施加电压,正电荷从正极注入,负电荷从负极注入,在电场作用下正负电荷在有机层中迁移相遇复合发光。由于oled器件具有亮度高、响应快、视角宽、工艺简单、可柔性化等优点,在新型显示技术领域和新型照明技术领域备受关注。目前,该技术已被广泛应用于新型照明灯具、智能手机及平板电脑等产品的显示面板,进一步还将向电视等大尺寸显示产品应用领域扩展,是一种发展快、技术要求高的新型显示技术。
[0003]
随着oled在照明和显示两大领域的不断推进,人们对于其核心材料的研究也更加关注。这是因为一个效率好、寿命长的oled器件通常是器件结构以及各种有机材料的优化搭配的结果,这就为化学家们设计开发各种结构的功能化材料提供了极大的机遇和挑战。常见的功能化有机材料有:空穴注入材料、空穴传输材料、空穴阻挡材料、电子注入材料、电子传输材料、电子阻挡材料以及发光主体材料和发光客体(染料)等。
[0004]
为了制备驱动电压更低、发光效率更好、器件使用寿命更长的oled发光器件,实现oled器件的性能不断提升,不仅需要对oled器件结构和制作工艺进行创新,更需要对oled器件中的光电功能材料不断研究和创新,以制备出具有更高性能的功能材料。基于此,oled材料界一直致力于开发新的有机电致发光材料以实现器件低启动电压、高发光效率和更优的使用寿命。
[0005]
到目前为止,现有的oled光电功能材料的发展还远远落后于面板制造企业对oled材料的要求,因此开发性能更好的有机功能材料满足当前产业发展需求显得尤为紧迫。
技术实现要素:
[0006]
本发明的主要目的在于提供一种化合物、其应用及包含其的有机电致发光器件,以解决现有技术中有机光电功能材料的启动电压高、发光效率低的问题。
[0007]
为了实现上述目的,根据本发明的一个方面,提供了一种化合物,该化合物具有如下式i所示结构:
[0008]
其中,l为单键、取代或未取代的c6~c
30
的亚芳基、取代或未取代的c3~c
30
的杂亚芳基;ar为取代或未取代的c6~c
30
的芳基、取代或未取代的c3~c
30
的杂芳基,m为1~6中的任意一个整数;r1、r2各自独立地选自取代或未取代的c1~c
20
的烷基、取代或未取代的c1~
c
12
的烷氧基、取代或未取代的c3~c
20
的环烷基、取代或未取代的c1~c6的醚基、取代或未取代的c6~c
30
的芳基、取代或未取代的c2~c
30
杂环烷基、取代或未取代的c3~c
30
的杂芳基,且r1、r2以单键的方式连接在母核上,n1和n2为0~4中的任意一个整数;g为c6~c
60
的多环亚环烷基,多环亚环烷基与式i中位于其两侧的两个苯基连接,且连接位为一个季碳原子;当上述各基团存在取代基时,取代基分别独立地选自卤素、c1~c
10
的烷基、c3~c
10
的环烷基、c1~c6的烷氧基、c1~c6的硫代烷氧基、c6~c
30
的芳基、c3~c
30
杂芳基中的一种或多种,式i中,“—”划过的环结构的表达方式表示连接位点于该环结构上任意能够成键的位置。
[0009]
根据本发明的另一方面,提供了一种上述任一种的化合物的应用,该应用为化合物在有机电致发光器件中作为空穴传输材料。
[0010]
根据本发明的另一方面,提供了一种有机电致发光器件,包括阳极和阴极以及设置在阳极和阴极之间的多层有机层,至少一个有机层中设置有化合物,至少一种化合物为上述任一种的化合物。
[0011]
应用本发明的技术方案,将上述的化合物作为有机电致发光器件的空穴传输层或电子阻挡层时,相比现有技术,能够进一步降低驱动电压、提高发光效率和延长使用寿命。具体地,上述化合物应用于器件时,上述性能的提升可能归结于多环环烷基在亚甲基上的占位导致结构中三芳胺分子间排列发生特定的重新排列,而基于该排列的无定型薄膜无论在空穴传输性能及稳定性均有大幅提升。
具体实施方式
[0012]
需要说明的是,在不冲突的情况下,本申请中的实施例及实施例中的特征可以相互组合。下面将结合实施例来详细说明本发明。
[0013]
同本领域的常规表达方式,以下结构式中“—”划过的环结构的表达方式表示连接位点于该环结构上任意能够成键的位置;“*”代表基团的接入位置。在本发明中,c6~c
30
的取代或未取代的亚芳基以及c6~c
30
取代或未取代的芳基中c6~c
30
表示基团中碳原子数目,优选碳原子数目为3、5、8、10、12、15、18、20、23、25、28或30;优选c3~c
30
的取代或未取代的亚杂芳基和c3~c
30
取代或未取代的杂芳基中碳原子数目为3、5、8、10、12、15、18、20、23、25、28或30;优选c1~c
20
的烷基中碳原子数为1、3、5、8、10、12、15、18或20。同样其他碳原子数范围的限定也表示所述基团的碳原子数可以取到所述数值范围内的任何一个整数。除非特殊说明,一般而言该碳原子数不包括取代基的碳原子数。本发明中,对于化学元素的表述包含化学性质相同的同位素的概念,例如“氢”的表述,也包括化学性质相同的“氘”、“氚”的概念。本发明中的杂原子,通常选自n、o、s。
[0014]
如本申请背景技术所分析的,现有技术的有机光电功能材料存在启动电压高、发光效率低的问题,为了解决该问题,本申请提供了一种化合物,该化合物具有如下式i所示结构:
[0015][0016]
其中,l为单键、取代或未取代的c6~c
30
的亚芳基、取代或未取代的c3~c
30
的杂亚
芳基;ar为取代或未取代的c6~c
30
的芳基、取代或未取代的c3~c
30
的杂芳基,m为1~6中的任意一个整数;r1、r2各自独立地选自取代或未取代的c1~c
20
的烷基、取代或未取代的c1~c
12
的烷氧基、取代或未取代的c3~c
20
的环烷基、取代或未取代的c1~c6的醚基、取代或未取代的c6~c
30
的芳基、取代或未取代的c2~c
30
杂环烷基、取代或未取代的c3~c
30
的杂芳基,且r1、r2以单键的方式连接在母核上,n1和n2为0~4中的任意一个整数;g为c6~c
60
的多环亚环烷基,且所述多环亚环烷基与所述式i中位于其两侧的两个苯基连接,且连接位为一个季碳原子,当上述各基团存在取代基时,取代基分别独立地选自c1~c
10
的烷基、c3~c
10
的环烷基、c1~c6的烷氧基、c1~c6的硫代烷氧基、c6~c
30
的芳基、c3~c
30
杂芳基中的一种或多种,式i中,“—”划过的环结构的表达方式表示连接位点于该环结构上任意能够成键的位置。
[0017]
将本申请上述的化合物作为有机电致发光器件的空穴传输层或电子阻挡层时,相比现有技术,能够进一步降低驱动电压、提高发光效率和延长使用寿命。具体地,上述化合物应用于器件时,上述性能的提升可能归结于多环环烷基在亚甲基上的占位导致结构中三芳胺分子间排列发生特定的重新排列,而基于该排列的无定型薄膜无论在空穴传输性能及稳定性均有大幅提升。
[0018]
在一种实施例中,优选上述式i中的g为c6~c
20
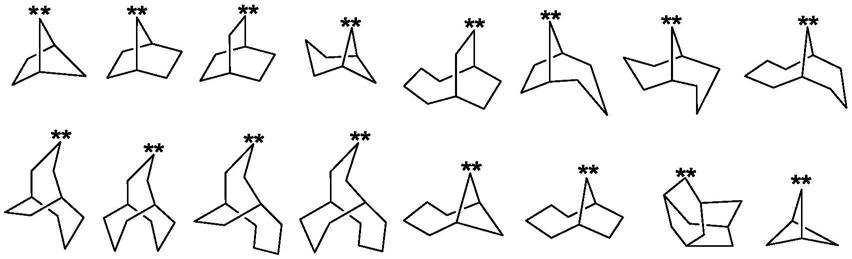
的多环亚环烷基,优选所述g为桥环的多环亚环烷基,进一步优选g选自以下多环亚环烷基之一:
[0019][0020]
其中,“*”代表基团的接入位置。
[0021]
优选地,上述ar为取代或未取代的c6~c
20
的芳基、取代或未取代的c5~c
20
的杂芳基。
[0022]
在另一种实施例中,优选上述r1、r2各自独立地选自取代或未取代的c1~c
10
的烷基、取代或未取代的c3~c6的醚基、取代或未取代的c3~c
10
的环烷基、取代或未取代的c6~c
20
的芳基、取代或未取代的c5~c
20
的杂芳基中的任意一种。
[0023]
上述取代或未取代的c1~c
20
烷基中的烷基优选为c1~c
10
的烷基,更优选为c1~c6的烷基,进一步优选为甲基、乙基、正丙基、异丙基、正丁基、正己基、正辛基、异丁基、叔丁基、环戊基或环己基。取代或未取代的c3~c
30
环烷基中的环烷基为c3~c
10
的环烷基,优选为环丙基、环丁基、环戊基或环己基。
[0024]
上述ar中或r1、r2中,c6~c
20
的芳基为由苯基、联苯基、三联苯基、萘基、蒽基、菲基、茚基、芴基及其衍生基团、荧蒽基、三亚苯基、芘基、苝基、基和并四苯基所组成的组中的任意一个基团,优选联苯基包括2-联苯基、3-联苯基和4-联苯基,优选三联苯基包括对-三联苯基-4-基、对-三联苯基-3-基、对-三联苯基-2-基、间-三联苯基-4-基、间-三联苯基-3-基和间-三联苯基-2-基;优选萘基包括1-萘基和2-萘基;优选蒽基包括1-蒽基、2-蒽基和9-蒽基;芴基包括1-芴基、2-芴基、3-芴基、4-芴基和9-芴基;优选芴基衍生基团包括9,9
’-
二
甲基芴基、9,9
’-
螺二芴基和苯并芴基;优选芘基包括1-芘基、2-芘基和4-芘基;优选并四苯基包括1-并四苯基、2-并四苯基和9-并四苯基;优选c5~c
20
杂芳基为呋喃基、噻吩基、吡咯基、苯并呋喃基、苯并噻吩基、异苯并呋喃基、吲哚基、二苯并呋喃基、二苯并噻吩基、咔唑基及其衍生基团组成的组中的任意一种,优选咔唑基衍生基团包括9-苯基咔唑基、9-萘基咔唑苯并咔唑基、二苯并咔唑基和吲哚并咔唑基。
[0025]
在一种实施例中,上述取代基分别独立地选自c1~c5的烷基、c1~c5的烷氧基、c6~c
20
的非稠环芳基、c5~c
20
非稠环杂芳基中的一种或多种。选用上述非稠环芳基或上述非稠环杂芳基作为取代基,可以降低芳环共轭程度有利于提高材料的三线态能级。优选上述取代基分别独立地选自氟、氯、溴、甲基、乙基、正丙基、异丙基、正丁基、正己基、正辛基、异丁基、叔丁基、环戊基、环己基、环丙基、环丁基、环戊基、环己基、四氢呋喃、吡咯烷、四氢噻吩、苯基、联苯基、三联苯基、2-联苯基、4-联苯基、对-三联苯基-4-基、对-三联苯基-3-基、对-三联苯基-2-基、间-三联苯基-4-基、间-三联苯基-3-基和间-三联苯基-2-基中的任意一种。
[0026]
上述r1、r2各自独立地为苯基、联苯基、萘基、芘基、9,9
’-
二甲基芴基、苯并芴基、二苯并呋喃基、二苯并噻吩基和9-苯基咔唑基中的任意一种,优选r1、r2各自独立地为苯基、联苯基、9,9
’-
二甲基芴基、二苯并呋喃基、二苯并噻吩基和9-苯基咔唑基中的任意一种。
[0027]
此外,优选上述n1和n2各自独立地为0或1或2。
[0028]
更为优选上述ar选自以下基团中的任意一种:
[0029]
[0030][0030]
其中,“—”划过的环结构的表达方式表示连接位点于该环结构上任意能够成键的位置,“*”代表基团的接入位置。
[0031]
上述m的数量可以选自1至6中的任意一个整数,优选为1。
[0032]
此外,优选l为单键。
[0033]
经过试验,优选上述化合物具有如式ii所示的结构:式ii,在一种实施例中,式ii中的l优选为单键。
[0034]
更优选上述化合物具有以下p1~p248中任意一个结构式所示结构:
[0035]
[0036]
[0037]
[0038]
[0039]
[0040]
[0041]
[0042]
[0043]
[0044]
[0045]
[0046]
[0047]
[0048]
[0049]
[0050]
[0051]
[0052][0053]
本申请的有机光电化合物的合成路线可以参考如下的合成路径:
[0054][0055]
基于以上化合物的合成路线和思路,本领域人员能够获得取代基为ar的式i所示化合物。
[0056]
在本申请另一种典型的实施方式中,提供了一种上述任一种的化合物的应用,该应用为所述化合物在有机电致发光器件中作为空穴传输材料。
[0057]
此外,本发明的化合物可以应用于有机电子器件中的空穴传输或电子阻挡层,该有机电子元器件可举出例如有机电致发光器件、照明元件、有机薄膜晶体管、有机场效应晶体管、有机薄膜太阳能电池、信息标签、电子人工皮肤片材、片材型扫描器等大面积传感器、电子纸及有机el面板等。
[0058]
在本申请另一种典型的实施方式中,还提供了一种有机电致发光器件,包括阳极和阴极以及设置在阳极和阴极之间的多层有机层,至少一个有机层中设置有有机光电化合物,至少一种有机光电化合物为上述任一种的化合物。将本申请的化合物应用于有机电致发光器件中时,可以降低器件的开启电压、提高光电效率并且延长器件寿命。
[0059]
在一种实施例中,上述有机层包括空穴传输层和/或电子阻挡层,本申请的化合物设置在空穴传输层和/或电子阻挡层中。
[0060]
此外,上述多层有机功能材料层还可以包括依次远离阳极设置的空穴传输层、空穴注入层、电子阻挡层、发光层、空穴阻挡层、电子注入层、电子传输层。
[0061]
接下来,对有机电致发光器件进行详细说明。
[0062]
有机电致发光器件包括第一电极和第二电极,以及位于电极之间的有机材料层。该有机材料层又可以分为多个区域。比如,该有机材料层可以包括空穴传输区、发光层、电子传输区。
[0063]
在具体实施例中,在第一电极下方或者第二电极上方可以使用基板。基板均为具有机械强度、热稳定性、防水性、透明度优异的玻璃或聚合物材料。此外,作为显示器用的基板上也可以带有薄膜晶体管(tft)。
[0064]
第一电极可以通过在基板上溅射或者沉积用作第一电极的材料的方式来形成。当第一电极作为阳极时,可以采用铟锡氧(ito)、铟锌氧(izo)、二氧化锡(sno2)、氧化锌(zno)等氧化物透明导电材料和它们的任意组合。第一电极作为阴极时,可以采用镁(mg)、银(ag)、铝(al)、铝-锂(al-li)、钙(ca)、镁-铟(mg-in)、镁-银(mg-ag)等金属或合金以及它们之间的任意组合。
[0065]
有机材料层可以通过真空热蒸镀、旋转涂敷、打印等方法形成于电极之上。用作有机材料层的化合物可以为有机小分子、有机大分子和聚合物以及它们的组合。
[0066]
空穴传输区位于阳极和发光层之间。空穴传输区可以为单层结构的空穴传输层(htl),包括只含有一种化合物的单层空穴传输层和含有多种化合物的单层空穴传输层。空穴传输区也可以为包括空穴注入层(hil)、空穴传输层(htl)、电子阻挡层(ebl)中的至少一层的多层结构。
[0067]
当空穴传输区的空穴传输层选自上述所述的化合物之一或任意组合时,空穴传输区的电子阻挡层可以没有,也可以有且选自、但不限于酞菁衍生物如cupc、导电聚合物或含
导电掺杂剂的聚合物如聚苯撑乙烯、聚苯胺/十二烷基苯磺酸(pani/dbsa)、聚(3,4-乙撑二氧噻吩)/聚(4-苯乙烯磺酸盐)(pedot/pss)、聚苯胺/樟脑磺酸(pani/csa)、聚苯胺/聚(4-苯乙烯磺酸盐)(pani/pss)、芳香胺衍生物如下面ht-1至ht-34所示的化合物;或者其任意组合。当空穴传输区的空穴传输层选自、但不限于酞菁衍生物如cupc、导电聚合物或含导电掺杂剂的聚合物如聚苯撑乙烯、聚苯胺/十二烷基苯磺酸(pani/dbsa)、聚(3,4-乙撑二氧噻吩)/聚(4-苯乙烯磺酸盐)(pedot/pss)、聚苯胺/樟脑磺酸(pani/csa)、聚苯胺/聚(4-苯乙烯磺酸盐)(pani/pss)、芳香胺衍生物如下面ht-1至ht-34所示的化合物,或者其任意组合时;空穴传输区的电子阻挡层选自上述所述的化合物之一或任意组合。
[0068]
[0069][0070]
空穴传输区及空穴注入区的材料可以选自、但不限于本发明所述化合物及上述化合物;或者其任意组合。其中,空穴注入层可以是单一化合物材料,也可以是多种化合物的组合。例如,空穴注入层可以采用上述本发明中的化合物中一种或多种化合物,或者采用下述hi1-hi3中的一种或多种化合物;也可以采用化合物中的一种或多种化合物掺杂下述hi1-hi3中的一种或多种化合物。
[0071][0072]
发光层包括可以发射不同波长光谱的发光染料(即掺杂剂,dopant),还可以同时包括主体材料(host)。发光层可以是发射红、绿、蓝等单一颜色的单色发光层。多种不同颜色的单色发光层可以按照像素图形进行平面排列,也可以堆叠在一起而形成彩色发光层。当不同颜色的发光层堆叠在一起时,它们可以彼此隔开,也可以彼此相连。发光层也可以是能同时发射红、绿、蓝等不同颜色的单一彩色发光层。
[0073]
根据不同的技术,发光层材料可以采用荧光电致发光材料、磷光电致发光材料、热活化延迟荧光发光材料等不同的材料。在一个oled器件中,可以采用单一的发光技术,也可以采用多种不同的发光技术的组合。这些按技术分类的不同发光材料可以发射同种颜色的光,也可以发射不同种颜色的光。
[0074]
在本发明的一方面,发光层采用磷光电致发光的技术。其发光层主体材料选自、但不限于gph-1至gph-80中的一种或多种的组合。
[0075]
[0076]
[0077][0078]
在本发明的一方面,发光层采用磷光电致发光的技术。其发光层磷光掺杂剂可以选自、但不限于以下所罗列的gpd-1至gpd-47的一种或多种的组合。
[0079]
[0080]
[0081][0082]
其中d为氘。
[0083]
在本发明的一方面,发光层采用磷光电致发光的技术。其发光层磷光掺杂剂可以选自、但不限于以下所罗列的rpd-1至rpd-28的一种或多种的组合。
[0084]
[0085][0086]
在本发明的一方面,发光层采用磷光电致发光的技术。其发光层磷光掺杂剂可以选自、但不限于以下所罗列的ypd-1—ypd-11的一种或多种的组合。
[0087][0088]
oled有机材料层还可以包括发光层与阴极之间的电子传输区。电子传输区可以为单层结构的电子传输层(etl),包括只含有一种化合物的单层电子传输层和含有多种化合物的单层电子传输层。电子传输区也可以为包括电子注入层(eil)、电子传输层(etl)、空穴阻挡层(hbl)中的至少一层的多层结构。
[0089]
本发明的一方面,电子传输层材料可以选自、但不限于以下所罗列的et-1至et-57的一种或多种的组合。
[0090]
[0091]
[0092][0093]
器件中还可以包括位于电子传输层与阴极之间的电子注入层,电子注入层材料包括但不限于以下罗列的一种或多种的组合:liq、lif、nacl、csf、li2o、cs2co3、bao、na、li和ca。
[0094]
下面通过具体实施例来进一步说明本发明的技术方案。本领域技术人员应该明了,所述实施例仅仅是帮助理解本发明,不应视为对本发明的具体限制。
[0095]
本发明中如下合成实施例中所用溶剂和试剂,例如芳基溴代物、2-溴-9,9
’-
二甲基芴、2-溴代二苯并呋喃、2-溴代二苯并噻吩、、4-溴联苯、[1,1
’-
双(二苯基膦)二茂铁]二氯化钯、三(二亚苄基丙酮)二钯、甲苯、石油醚、正己烷、二氯甲烷、丙酮、硫酸钠、乙酸乙酯、乙醇、三特丁基膦、叔丁基醇钾/钠等化学试剂,均可以从国内化工产品市场购买或定制,例如购买自国药集团试剂公司、sigma-aldrich公司、百灵威试剂公司,中间体m由郑州海阔、百灵威提供。另外,本领域技术人员也可以通过公知方法合成。
[0096]
合成实施例1:化合物p1的合成
[0097][0098]
在500ml单口瓶中,加入15g(50mmol)m1、7.8g(50mmol)溴苯、0.9g(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入
甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p1所示的化合物,m/z理论值:377,m/z实测值:378。
[0099]
合成实施例2:化合物p2的合成
[0100][0101]
在500ml单口瓶中,加入15g(50mmol)m1、10.5g(50mmol)1-溴萘、0.9g(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p2所示的化合物,m/z理论值:427,m/z实测值:428。
[0102]
合成实施例3:化合物p3的合成
[0103][0104]
在500ml单口瓶中,加入15g(50mmol)m1、10.5g(50mmol)2-溴萘、0.9g(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p3所示的化合物,m/z理论值:427,m/z实测值:428。
[0105]
合成实施例4:化合物p6合成
[0106][0107]
在500ml单口瓶中,加入15g(50mmol)m1、11.5g(50mmol)3-溴联苯、0.9g(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完
毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p6所示的化合物,m/z理论值:453,m/z实测值:454。
[0108]
合成实施例5:化合物p9合成
[0109][0110]
在500ml单口瓶中,加入15g(50mmol)m1、15.4g(50mmol)3,5-二苯基溴苯、0.9g(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p9所示的化合物,m/z理论值:529,m/z实测值:530。
[0111]
合成实施例6:化合物p17合成
[0112][0113]
在500ml单口瓶中,加入15g(50mmol)m1、13.1g(50mmol)2-溴二苯并噻吩、0.9g(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p17所示的化合物,.,m/z理论值:483,m/z实测值:484。
[0114]
合成实施例7:化合物p18合成
[0115]
[0116]
在500ml单口瓶中,加入15g(50mmol)m1、12.3g(50mmol)2-溴二苯并呋喃、0.9g(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p18所示的化合物,m/z理论值:468,m/z实测值:469。
[0117]
合成实施例8:化合物p19合成
[0118][0119]
在500ml单口瓶中,加入15g(50mmol)m1、13.6g(50mmol)2-溴-9,9-二甲基芴、0.9g(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p19所示的化合物,m/z理论值:493,m/z实测值:494。
[0120]
合成实施例9:化合物p25合成
[0121][0122]
在500ml单口瓶中,加入15g(50mmol)m1、13.1g(50mmol)4-溴二苯并噻吩、0.9g(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p25所示的化合物,m/z理论值:483,m/z实测值:484。
[0123]
合成实施例10:化合物p26合成
[0124]
[0125]
在500ml单口瓶中,加入15g(50mmol)m1、12.3g(50mmol)4-溴二苯并呋喃、0.9g(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p26所示的化合物,m/z理论值:468,m/z实测值:469。
[0126]
合成实施例11:化合物p45合成
[0127][0128]
在500ml单口瓶中,加入16g(50mmol)m2、7.8g(50mmol)溴苯、0.9g(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p45所示的化合物,m/z理论值:393,m/z实测值:394。
[0129]
合成实施例12:化合物p50合成
[0130][0131]
在500ml单口瓶中,加入16g(50mmol)m2、11.5g(50mmol)3-溴联苯、0.9g(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、714.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p50所示的化合物,m/z理论值:469,m/z实测值:470。
[0132]
合成实施例13:化合物p53合成
[0133][0134]
在500ml单口瓶中,加入16g(50mmol)m2、15.4g(50mmol)3,5-二苯基溴苯、0.9g
(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p53所示的化合物,m/z理论值:545,m/z实测值:546。
[0135]
合成实施例14:化合物p61合成
[0136][0137]
在500ml单口瓶中,加入16g(50mmol)m2、13.1g(50mmol)2-溴二苯并噻吩、0.9g(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p61所示的化合物,m/z理论值:499,m/z实测值:500。
[0138]
合成实施例15:化合物p62合成
[0139][0140]
在500ml单口瓶中,加入16g(50mmol)m2、12.3g(50mmol)2-溴二苯并呋喃、0.9g(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p62所示的化合物,m/z理论值:483,m/z实测值:484。
[0141]
合成实施例16:化合物p105合成
[0142][0143]
在500ml单口瓶中,加入11.5g(50mmol)m3、13.1g(50mmol)2-溴二苯并噻吩、0.9g
(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p105所示的化合物,m/z理论值:416,m/z实测值:417。
[0144]
合成实施例17:化合物p106合成
[0145][0146]
在500ml单口瓶中,加入11.5g(50mmol)m3、12.3g(50mmol)2-溴二苯并呋喃、0.9g(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p106所示的化合物,m/z理论值:399,m/z实测值:400。
[0147]
合成实施例18:化合物p149合成
[0148][0149]
在500ml单口瓶中,加入13.8g(50mmol)m4、13.1g(50mmol)2-溴二苯并噻吩、0.9g(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p149所示的化合物,m/z理论值:458,m/z实测值:459。
[0150]
合成实施例19:化合物p150合成
[0151][0152]
在500ml单口瓶中,加入13.8g(50mmol)m4、12.3g(50mmol)2-溴二苯并呋喃、0.9g
(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p150所示的化合物,m/z理论值:442,m/z实测值:443。
[0153]
合成实施例20:化合物p177合成
[0154][0155]
在500ml单口瓶中,加入15g(50mmol)m5、12.3g(50mmol)2-溴二苯并呋喃、0.9g(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p177所示的化合物,m/z理论值:469,m/z实测值:470。
[0156]
合成实施例21:化合物p178合成
[0157][0158]
在500ml单口瓶中,加入12.3g(50mmol)m6、12.3g(50mmol)2-溴二苯并呋喃、0.9g(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p178所示的化合物,m/z理论值:416,m/z实测值:417。
[0159]
合成实施例22:化合物p179合成
[0160][0161]
在500ml单口瓶中,加入15g(50mmol)m7、12.3g(50mmol)2-溴二苯并呋喃、0.9g
(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p179所示的化合物,m/z理论值:469,m/z实测值:470。
[0162]
合成实施例23:化合物p180合成
[0163][0164]
在500ml单口瓶中,加入13g(50mmol)m8、12.3g(50mmol)2-溴二苯并呋喃、0.9g(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p180所示的化合物,m/z理论值:428,m/z实测值:429。
[0165]
合成实施例24:化合物p181合成
[0166][0167]
在500ml单口瓶中,加入16g(50mmol)m9、12.3g(50mmol)2-溴二苯并呋喃、0.9g(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p181所示的化合物,m/z理论值:483,m/z实测值:484。
[0168]
合成实施例25:化合物p182合成
[0169][0170]
在500ml单口瓶中,加入13g(50mmol)m10、12.3g(50mmol)2-溴二苯并呋喃、0.9g
(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p182所示的化合物,m/z理论值:427,m/z实测值:428。
[0171]
合成实施例26:化合物p183合成
[0172][0173]
在500ml单口瓶中,加入15g(50mmol)m11、12.3g(50mmol)2-溴二苯并呋喃、0.9g(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p183所示的化合物,m/z理论值:497,m/z实测值:498。
[0174]
合成实施例27:化合物p184合成
[0175][0176]
在500ml单口瓶中,加入15g(50mmol)m12、12.3g(50mmol)2-溴二苯并呋喃、0.9g(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p184所示的化合物,m/z理论值:469,m/z实测值:470.
[0177]
合成实施例28:化合物p185合成
[0178][0179]
在500ml单口瓶中,加入13.5g(50mmol)m13、12.3g(50mmol)2-溴二苯并呋喃、0.9g
(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p185所示的化合物,m/z理论值:441,m/z实测值:442.
[0180]
合成实施例29:化合物p186合成
[0181][0182]
在500ml单口瓶中,加入13.5g(50mmol)m14、12.3g(50mmol)2-溴二苯并呋喃、0.9g(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p186所示的化合物,m/z理论值:441,m/z实测值:442.
[0183]
合成实施例30:化合物p187合成
[0184][0185]
在500ml单口瓶中,加入14.4g(50mmol)m15、12.3g(50mmol)2-溴二苯并呋喃、0.9g(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p187所示的化合物,m/z理论值:455,m/z实测值:456.
[0186]
合成实施例31:化合物p188合成
[0187]
[0188]
在500ml单口瓶中,加入14.4g(50mmol)m16、12.3g(50mmol)2-溴二苯并呋喃、0.9g(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p188所示的化合物,m/z理论值:455,m/z实测值:456.
[0189]
合成实施例32:化合物p189合成
[0190][0191]
在500ml单口瓶中,加入18.5g(50mmol)m17、13.6g(50mmol)2-溴-9,9-二甲基芴、0.9g(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p189所示的化合物,m/z理论值:569,m/z实测值:570。
[0192]
合成实施例33:化合物p190合成
[0193][0194]
在500ml单口瓶中,加入22.5g(50mmol)m18、13.6g(50mmol)2-溴-9,9-二甲基芴、0.9g(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p190所示的化合物,m/z理论值:645,m/z实测值:646。
[0195]
合成实施例34:化合物p191合成
[0196][0197]
在500ml单口瓶中,加入26.5g(50mmol)m19、13.6g(50mmol)2-溴-9,9-二甲基芴、0.9g(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p191所示的化合物,m/z理论值:721,m/z实测值:722。
[0198]
合成实施例35:化合物p192合成
[0199][0200]
在500ml单口瓶中,加入18.5g(50mmol)m20、13.6g(50mmol)2-溴-9,9-二甲基芴、0.9g(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p192所示的化合物,m/z理论值:586,m/z实测值:587。
[0201]
合成实施例36:化合物p193合成
[0202][0203]
在500ml单口瓶中,加入23.5g(50mmol)m21、13.6g(50mmol)2-溴-9,9-二甲基芴、0.9g(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,
向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p193所示的化合物,m/z理论值:661,m/z实测值:662。
[0204]
合成实施例37:化合物p194合成
[0205][0206]
在500ml单口瓶中,加入27.2g(50mmol)m22、13.6g(50mmol)2-溴-9,9-二甲基芴、0.9g(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p194所示的化合物,m/z理论值:737,m/z实测值:738。
[0207]
合成实施例38:化合物p12的合成
[0208][0209]
在500ml单口瓶中,加入15g(50mmol)m1、18g(50mmol)1-溴-2-(1-萘基)联苯、0.9g(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p12所示的化合物,m/z理论值:579,m/z实测值:580。
[0210]
合成实施例39:化合物p38的合成
[0211][0212]
在500ml单口瓶中,加入15g(50mmol)m1、16g(50mmol)4-溴-9-苯基-咔唑、0.9g(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应
5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p38所示的化合物,m/z理论值:542,m/z实测值:543。
[0213]
合成实施例40:化合物p128合成
[0214][0215]
在500ml单口瓶中,加入12.3g(50mmol)m6、15g(50mmol)2-溴苯并萘并呋喃、0.9g(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p128所示的化合物,m/z理论值:449,m/z实测值:450。
[0216]
合成实施例41:化合物p213合成
[0217][0218]
在500ml单口瓶中,加入15g(50mmol)m1、21.8g(50mmol)z1、0.9g(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p213所示的化合物,m/z理论值:633,m/z实测值:634。
[0219]
合成实施例42:化合物p218合成
[0220]
[0221]
在500ml单口瓶中,加入30g(100mmol)m1、13g(50mmol)z2、0.9g(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p218所示的化合物,m/z理论值:701,m/z实测值:702。
[0222]
合成实施例43:化合物p224合成
[0223][0224]
在500ml单口瓶中,加入30g(100mmol)m1、16.5g(50mmol)z3、0.9g(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p224所示的化合物,m/z理论值:777,m/z实测值:778。
[0225]
合成实施例44:化合物p241合成
[0226][0227]
在500ml单口瓶中,加入32g(100mmol)m2、18g(50mmol)z4、0.9g(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p241所示的化合物,m/z理论值:834,m/z实测值:835。
[0228]
合成实施例45:化合物p245合成
[0229][0230]
在500ml单口瓶中,加入15.8g(50mmol)m2、10.3g(50mmol)z5、0.9g(1mmol)三(二亚苄基丙酮)二钯(即pd2(dba)3)、0.5ml三特丁基膦((t-bu)3p)、300ml甲苯(toluene)、14.4g(150mmol)叔丁醇钠(naobu-t),抽真空换氮气3次,反应升温至110℃反应5h,反应完毕,停止反应,得到反应液。将反应液冷却至室温,对反应液分液,浓缩有机相,向浓缩后的有机相加入甲醇搅拌1h,抽滤得到淡黄色粉末,为结构式p245所示的化合物,m/z理论值:443,m/z实测值:444。
[0231]
器件实施例1
[0232]
本实施例中有机电致发光器件制备过程如下:
[0233]
将涂布了ito透明导电层(作为阳极)的玻璃板在商用清洗剂中超声处理后,在去离子水中冲洗,在丙酮:乙醇混合溶剂中超声除油,在洁净环境下烘烤至完全除去水份,用紫外光和臭氧清洗,并用低能阳离子束轰击表面;
[0234]
把上述带有阳极的玻璃基片置于真空腔内,抽真空至1
×
10-5
pa,在上述阳极层膜上真空蒸镀hi-3作为空穴注入层,蒸镀速率为0.1nm/s,蒸镀膜厚为10nm;
[0235]
在空穴注入层之上真空蒸镀合成实施例1制备的化合物p1作为器件的空穴传输层,蒸镀速率为0.1nm/s,蒸镀总膜厚为80nm;
[0236]
在空穴传输层之上,真空蒸镀ht-14作为器件的电子阻挡层,蒸镀速率为0.1nm/s,蒸镀总膜厚为80nm;
[0237]
在电子阻挡层之上真空蒸镀器件的发光层,发光层包括主体材料和染料材料,利用多源共蒸的方法,调节主体材料gph-59蒸镀速率为0.1nm/s,染料rpd-8蒸镀速率为主体材料的3%进行比例设定,蒸镀总膜厚为30nm;
[0238]
在发光层之上真空蒸镀器件的电子传输层材料et-46和et-57,二者质量比为1:1,其蒸镀速率为0.1nm/s,蒸镀总膜厚为30nm;
[0239]
在电子传输层(etl)上真空蒸镀厚度为0.5nm的lif作为电子注入层,厚度为150nm的al层作为器件的阴极。
[0240]
器件实施例2至7
[0241]
各器件实施例中有机电致发光器件制备过程与器件实施例1中相同,区别仅在于将化合物p1替换为表1中对应的化合物作为空穴传输层材料。
[0242]
器件比较例1
[0243]
在该比较例中,有机电致发光器件制备过程与实施例1中相同,区别仅在于,将化合物p1替换为具有如下结构iii的化合物作为空穴传输材料:
[0244][0245]
对器件实施例1-7和器件比较例1制备的有机电致发光器件进行如下性能测定:
[0246]
在同样亮度下,使用数字源表及亮度计测定器件实施例1-7和器件比较例1制备得到的有机电致发光器件的驱动电压和电流效率以及器件的寿命。具体而言,以每秒0.1v的速率提升电压,测定当有机电致发光器件的亮度达到5000cd/m2时的电压即驱动电压,同时测出此时的电流密度;亮度与电流密度的比值即为电流效率;lt95的寿命测试如下:使用亮度计在5000cd/m2亮度下,保持恒定的电流,测量有机电致发光器件的亮度降为4750cd/m2的时间,单位为小时。测定结果如表1所示。
[0247]
表1
[0248][0249][0250]
从表1结果可以看出,本发明的化合物用于有机电致发光器件的空穴传输材料时,器件亮度达到5000cd/m2时,驱动电压低至8.9v以下,电流效率高达13cd/a以上,lt95达到16h以上,可以有效的降低驱动电压、提高电流效率、延长器件使用寿命,是性能良好的空穴传输材料。
[0251]
而对比例1的化合物iii与实施例3中的化合物p3相比,区别仅在烷基部分的环己基变成多环基,该化合物用作有机电致发光器件的空穴传输材料时,器件的驱动电压为10.5v,电流效率为8.5cd/a,lt95寿命为6h,各项数据相较于实施例3均变差,这可能归结于化合物p3具有更好分子间更好的堆积进而具有更好的空穴传输性能。
[0252]
器件实施例8
[0253]
本实施例中有机电致发光器件制备与前述器件实施例1的区别在于:
[0254]
在空穴注入层之上真空蒸镀ht-4作为器件的空穴传输层,蒸镀速率为0.1nm/s,蒸镀总膜厚为80nm;在空穴传输层之上真空蒸镀化合物p2作为器件的电子阻挡层,蒸镀速率
为0.1nm/s,蒸镀总膜厚为5nm;在电子阻挡层之上真空蒸镀器件的发光层,发光层包括主体材料和染料材料,利用多源共蒸的方法,调节bfh-2作为主体材料,蒸镀速率为0.1nm/s,染料bfd-4蒸镀速率3%比例设定,蒸镀总膜厚为30nm;在发光层之上真空蒸镀器件的电子传输层材料et46形成电子传输层,其蒸镀速率为0.1nm/s,蒸镀总膜厚为30nm;在电子传输层(etl)上真空蒸镀厚度为0.5nm的lif作为电子注入层,厚度为150nm的al层作为器件的阴极。
[0255]
器件实施例9-15与器件实施例8的区别仅在于将空穴传输材料化合物p2替换为其他化合物,具体详见表2。器件比较例2
[0256]
在该比较例中,有机电致发光器件制备过程与器件实施例8中相同,区别仅在于,将化合物p2替换为具有如下结构iii的化合物作为电子阻挡材料:
[0257][0258]
对由上述器件实施例8-15以及器件比较例2过程制备的有机电致发光器件进行如下性能测定:
[0259]
在同样亮度下,使用数字源表及亮度计测定器件实施例8-15以及器件比较例2中制备得到的有机电致发光器件的驱动电压和电流效率以及器件的寿命。具体而言,以每秒0.1v的速率提升电压,测定当有机电致发光器件的亮度达到5000cd/m2时的电压即驱动电压,同时测出此时的电流密度;亮度与电流密度的比值即为电流效率;lt95的寿命测试如下:使用亮度计在5000cd/m2亮度下,保持恒定的电流,测量有机电致发光器件的亮度降为4750cd/m2的时间,单位为小时,测定结果如表2所示。
[0260]
表2
[0261]
[0262]
由表2中数据可以看出,本发明的化合物用于有机电致发光器件的电子阻挡层材料时,器件亮度达到5000cd/m2时,驱动电压低至8.5v以下,电流效率高达11cd/a以上,lt95达到17h以上,可以有效的降低驱动电压、提高电流效率、延长器件使用寿命,是性能良好的电子阻挡层材料。
[0263]
而器件比较例2的化合物iii与器件实施例9中的化合物p3相比,区别仅在烷基部分的环己基变成多环基,该化合物用作有机电致发光器件的空穴传输材料时,器件的驱动电压为9.8v,电流效率为10.8cd/a,lt95寿命为12h,各项数据相较于器件实施例9均变差,这可能归结于化合物p3具有更好分子间更好的堆积进而具有更好的空穴传输性能。
[0264]
器件实施例16
[0265]
本实施例中有机电致发光器件制备过程如下:
[0266]
将涂布了ito(50nm)透明导电层的玻璃板在商用清洗剂中超声处理,在去离子水中冲洗,在丙酮∶乙醇混合溶剂(体积比1∶1)中超声除油,在洁净环境下烘烤至完全除去水份,用紫外光和臭氧清洗,并用低能阳离子束轰击表面。
[0267]
把上述带有阳极的玻璃基片置于真空腔内,抽真空至1
×
10-5
,在上述阳极层膜上真空蒸镀2-tnata(4,4',4"-三[n,n-(2-萘基)-苯基氨基]三苯胺),形成厚度为60nm的空穴注入层;在空穴注入层之上真空蒸镀化合物npb,形成厚度为20nm的空穴传输层,蒸镀速率为0.1nm/s。
[0268]
在上述空穴传输层上形成电致发光层,具体操作为:将作为发光层主体的化合物tdh1放置在真空气相沉积设备的小室中,将作为掺杂剂的化合物p213放置在真空气相沉积设备的另一室中,以不同的速率同时蒸发两种材料,化合物p213的浓度为20wt%,蒸镀总膜厚为30nm。
[0269]
在发光层之上真空蒸镀bphen形成厚膜为20nm的电子传输层,其蒸镀速率为0.1nm/s。
[0270]
在电子传输层上真空蒸镀0.5nm的lif作为电子注入层和厚度为150nm的al层作为器件的阴极。
[0271]
器件实施例17-20与器件实施例16的区别仅在于将空穴传输材料化合物p213替换为其他化合物,具体详见表3。
[0272]
器件比较例3
[0273]
在该比较例中,有机电致发光器件制备过程与器件实施例16中相同,区别仅在于,将化合物p213替换为具有如下结构iv的化合物作为发光掺杂剂:
[0274][0275]
对器件实施例16-20、器件比较例3得到的有机电致发光器件进行前文所述的性能测试,测试结果如表3所示。
[0276]
表3
[0277] 电子阻挡层材料要求亮度(cd/m2)电压(v)电流效率(cd/a)
比较例3iv1000.007.29.1实施例16p2131000.006.59.9实施例17p2181000.005.811.2实施例18p2241000.006.210.8实施例19p2411000.005.211实施例20p2451000.006.810.6
[0278]
由表3中数据可以看出,本发明的化合物用于有机电致发光器件的电子阻挡层材料时,器件亮度达到1000cd/m2时,驱动电压低至6.8v以下,电流效率高达9.9cd/a以上,相比对比例化合物制备器件,可以实现更蓝的发光。
[0279]
而器件比较例3的化合物iv与器件实施例20中的化合物p245相比,区别仅在烷基部分的环己基变成多环基,该化合物用作有机电致发光器件的掺杂剂时,器件的驱动电压为7.2v,电流效率为9.1cd/a,各项数据相较于器件实施例20均变差,这可能归结于化合物p245具有更高的三线态(高斯计算对比列及p245的三线态能级分别为2.52ev、2.67ev)、更优的分子间距离能够有效降低发光分子淬灭及分子具有更好的线性排列提高材料的折光系数从而提高器件光取出率。
[0280]
本发明通过上述实施例来说明本发明的化合物及其在oled器件上的应用,但本发明并不局限于上述实施例,即不意味着本发明必须依赖上述实施例才能实施。所属技术领域的技术人员应该明了,对本发明的任何改进,对本发明产品各原料的等效替换及辅助成分的添加、具体方式的选择等,均落在本发明的保护范围和公开范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1