改性发泡剂、微发泡聚丙烯材料及制备方法与流程
本发明涉及高分子材料领域,具体涉及一种改性发泡剂、微发泡聚丙烯材料及制备方法。
背景技术:
微发泡聚丙烯专用料是近年来兴起的一种改性料,具有制品收缩率低、成型时间短、减重、节省原料等优势,在汽车零部件、电子产品等领域获具有广泛的应用前景。偶氮二甲酰胺(ac)是生产微发泡聚丙烯最常用的发泡剂之一,中国专利申请201310727884和中国专利申请201710559553都在微发泡聚丙烯中使用了ac发泡剂。ac发泡剂在发泡过程中分解产物主要包括n2、co、co2、氨气,由于氨气的存在导致采用ac发泡剂制备的制品不能满足环保的要求,严重限制了ac发泡剂在聚丙烯中的使用。
如何通过对偶氮二甲酰胺进行改性,获得环保型聚丙烯材料,至关重要。
技术实现要素:
本发明旨在至少在一定程度上解决相关技术中的技术问题之一。为此,本发明的一个目的在于提出一种改性发泡剂、微发泡聚丙烯材料及其制备方法。
偶氮二甲酰胺(ac)发泡剂在发泡过程中分解产物主要包括n2、co、co2和氨气,由于氨气的存在导致采用ac发泡剂制备的制品不能满足环保的要求,严重限制了ac发泡剂在聚丙烯中的使用。
为此,本发明提出一种ac发泡剂的化学改性方法,通过引入带有环氧基团的脂肪酸缩水甘油酯,将ac发泡剂分子结构上的伯胺转变为仲胺,一方面降低了胺基的活性,另一方面,通过引入脂肪酸的长碳链,限制了氨气的生成,从根本上解决了ac发泡剂热分解产物中生成氨气的问题。
同时还利用上述改性发泡剂制备获得微发泡聚丙烯材料。即聚丙烯树脂为基体,通过添加改性发泡剂、成核剂,采用双螺杆挤出机熔融造粒而成。由此获得的微发泡聚丙烯材料无刺激性气体释放,泡孔尺寸均一,分布均匀,可广泛应用于汽车零部件、电子产品等领域的应用。
具体而言,本发明提供了如下技术方案:
根据本发明的第一方面,本发明提供了一种改性发泡剂,包括:经脂肪族缩水甘油酯改性的偶氮二甲酰胺。本发明所提供的改性发泡剂,通过引入带有环氧基团的脂肪酸缩水甘油酯,将ac发泡剂分子结构上的伯胺转变为仲胺,一方面降低了胺基的活性,另一方面,通过引入脂肪酸的长碳链,限制了氨气的生成,从根本上解决了ac发泡剂热分解产物中生成氨气的问题。从而使得本发明所提供的改性发泡剂更加环保。
进一步地,所述经脂肪族缩水甘油酯改性的偶氮二甲酰胺由偶氮二甲酰胺和脂肪族缩水甘油酯按照摩尔比为1:1.5~1:3制备而成。由此,可以将将脂肪链化学接枝到偶氮二甲酰胺分子结构上,获得经脂肪族缩水甘油酯改性的偶氮二甲酰胺。当脂肪族缩水甘油酯的量过少时,不足以反应掉足够的酰胺基,发泡剂分解时仍然生成较多氨气;当脂肪族缩水甘油酯的量过多时,会造成ac中两个酰胺基发生进一步反应的情况,造成产物分解温度过高,降低树脂加工过程中发泡剂的分解效率,减少发气量。
进一步地,所述脂肪族缩水甘油酯包括选自棕榈酸缩水甘油酯、油酸缩水甘油酯、亚油酸缩水甘油酯、亚麻酸缩水甘油酯、甲基丙烯酸缩水甘油酯中的至少一种。由此可以获得环保的经脂肪族缩水甘油酯改性的偶氮二甲酰胺。
根据本发明的第二方面,本发明提供了一种改性发泡剂的制备方法,所述改性发泡剂为本发明第一方面所述的改性发泡剂,所述制备方法包括:将偶氮二甲酰胺和脂肪族缩水甘油酯在溶剂中进行反应,以便获得所述改性发泡剂。
进一步地,所述反应温度为55~65摄氏度,所述反应时间为1~3小时,优选为2~3小时。在55~65摄氏度下,偶氮二甲酰胺和脂肪族缩水甘油酯反应完全,可以快速获得环保的氨气生成量较少的改性发泡剂。
进一步地,所述溶剂为二甲基甲酰胺或二甲基亚砜,所述偶氮二甲酰胺和所述脂肪族缩水甘油酯在所述溶剂中的固含量为10~30%。
根据本发明的第三方面,本发明提供了一种微发泡聚丙烯材料,包括:聚丙烯树脂,改性发泡剂,所述改性发泡剂为本发明第一方面所述的改性发泡剂或者根据本发明第二方面所述的方法制备的改性发泡剂;成核剂,抗氧剂,和润滑剂。本发明通过上述改性发泡剂制备微发泡聚丙烯材料,耐热温度高,获得的聚丙烯材料在熔融造粒过程中发泡剂不分解,塑料制品注塑成型过程中发泡稳定,无刺激性氨气产生,有效提高了微发泡聚丙烯专用料的环保特性。
进一步地,所述改性发泡剂与所述聚丙烯树脂的质量百分比为0.1~5%,优选为1~2%。
进一步地,所述成核剂与所述聚丙烯的质量百分比为1~30%,优选为1~10%。所述抗氧剂与所述聚丙烯树脂的质量百分比为0.2~1%,所述润滑剂与所述聚丙烯树脂的质量百分比为0.5~1%。
进一步地,所述成核剂选自滑石粉、3,4-二甲基亚苄基山梨醇或甲撑双(4,6-二叔丁基苯基)磷酸钠中的至少一种;所述抗氧剂选自1010、dltp、168中的至少一种;所述润滑剂选自聚乙烯蜡、硬脂酸钙、硬脂酸锌、ebs、石蜡的至少一种。
进一步地,所述聚丙烯树脂的熔融指数范围1~90g/10min。
根据本发明的第四方面,本发明提供了一种微发泡聚丙烯材料的制备方法,所述微发泡聚丙烯材料为本发明第三方面所述的微发泡聚丙烯材料,所述制备方法包括:将所述聚丙烯、改性发泡剂、成核剂、抗氧剂和润滑剂混合,进行熔融造粒,以便获得所述微发泡聚丙烯材料。由此可以制得各方面性能较好,且环保的微发泡聚丙烯材料。
进一步地,所述熔融造粒的温度为140~230℃。由此可以快速制备获得微发泡聚丙烯材料。
附图说明
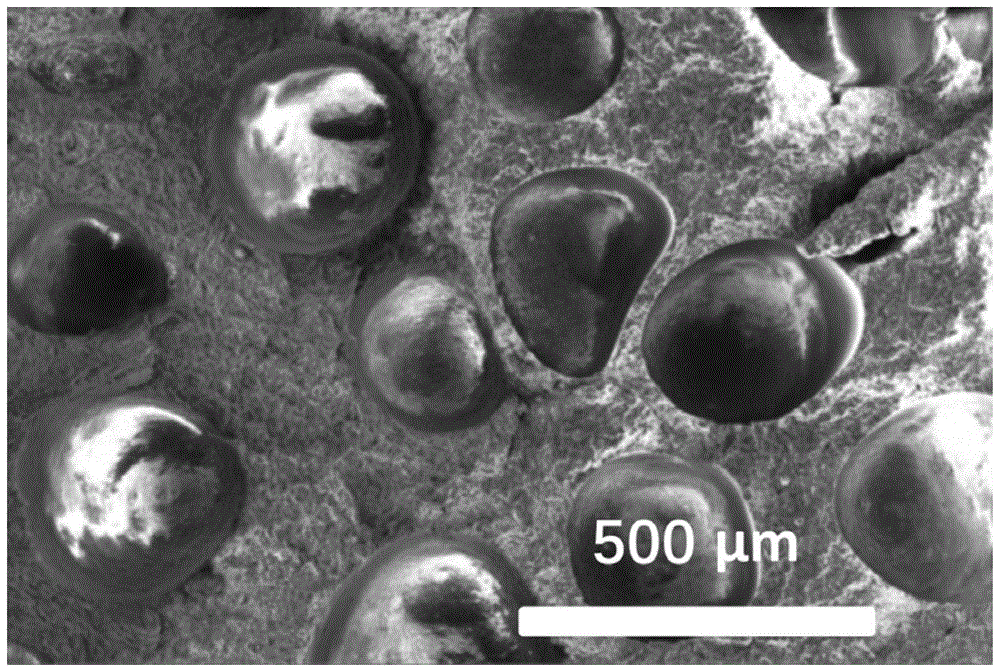
图1是根据本发明的实施例提供的微发泡聚丙烯注塑件截面的电镜图。
具体实施方式
在本发明的至少一些实施方式中,本发明提供了改性发泡剂的制备方法,包括:将偶氮二甲酰胺和脂肪族缩水甘油酯按照1:1.5~1:3摩尔比加入烧瓶中,溶剂为二甲基甲酰胺或二甲基亚砜,固含量为10~30%,置于水浴中60℃条件下,控制反应时间1~3小时,脱除溶剂后烘干,制得改性发泡剂。在本发明的至少一些实施方式中,所获得的改性发泡剂的分解温度为222摄氏度~230摄氏度,例如为224摄氏度~227摄氏度,所生成的气体的氨气含量小于1%。
利用上述方法制备的改性发泡剂可以用来制备微发泡聚丙烯材料,所制备的微发泡聚丙烯材料的耐热温度高,而且在熔融造粒过程中改性发泡剂不分解,塑料制品注塑成型过程中发泡稳定,无刺激性氨气产生,环保性能佳,符合现代社会对于环保的需求。
下面将结合实施例对本发明的方案进行解释。本领域技术人员将会理解,下面的实施例仅用于说明本发明,而不应视为限定本发明的范围。实施例中未注明具体技术或条件的,按照本领域内的文献所描述的技术或条件或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。
实施例1
实施例1提供了制备改性发泡剂的方法。
对比例
对比例为纯偶氮二甲酰胺发泡剂,通过tga(热重分析法)测试,改性发泡剂的分解温度为201℃。通过中华人民共和国化工行业标准hg2097-91排水法测试,改性发泡剂的发气量为217ml/g。通过吸收滴定法测试,生成气体中的氨气含量(v/v)为17.4%。
实验组1
实验组1通过下述方法制备改性发泡剂:
将偶氮二甲酰胺和油酸缩水甘油酯按照1:1.5摩尔比加入烧瓶中,溶剂为二甲基甲酰胺,固含量为10%,置于水浴中60℃条件下,反应2小时,然后脱除溶剂后烘干,制得改性发泡剂。
通过tga(热重分析法)测试,改性发泡剂的分解温度为224℃。通过中华人民共和国化工行业标准hg2097-91排水法测试,改性发泡剂的发气量为45ml/g。通过吸收滴定法测试,生成气体中的氨气含量(v/v)为0.6%。
实验组2
实验组2通过下述方法制备改性发泡剂:
将偶氮二甲酰胺和亚麻酸缩水甘油酯按照1:3摩尔比加入烧瓶中,溶剂为二甲基亚砜,固含量为30%,置于水浴中60℃条件下,反应3小时,然后脱除溶剂后烘干,制得改性发泡剂。
参照实验组1的方法,通过tga测试,实验组2所制得的改性发泡剂的分解温度为227℃。通过排水法测试,改性发泡剂的发气量为26ml/g。通过吸收滴定法测试,生成气体中的氨气含量为0。
实验组3
实验组3与实验组1不同的是,实验组3中偶氮二甲酰胺和亚油酸缩水甘油酯的摩尔比为1:1。
由此所获得的改性发泡剂分解气体中氨气含量达到4.2%。说明当脂肪族缩水甘油酯的比例过少时,不足以反应掉足够的酰胺基,发泡剂分解时仍然生成较多氨气。
实验组4
实验组4与实验组1不同的是,实验组4中偶氮二甲酰胺和棕榈酸缩水甘油酯的摩比比为1:4。
由此所获得的改性发泡剂发气量为3ml/g。说明当脂肪族缩水甘油酯的比例过多时,会造成ac中两个酰胺基都发生反应的情况,造成产物分解温度过高,在发泡过程中不分解,减少发气量。
实验组5
实验组5与实验组1不同的是,在烧瓶中的固体含量为40%。
由此所获得的改性发泡剂发气量为55ml/g,分解气体中氨气含量达到6.7%。说明当固体含量过高时,反应发生不完全,部分ac没有与脂肪酸缩水甘油酯发生反应。
实验组6
实验组6与实验组1不同的是,在水浴中温度40℃下反应。
由此所获得的改性发泡剂发气量为61ml/g,分解气体中氨气含量达到8.8%。说明当反应温度过低时,反应发生不完全,部分ac没有与脂肪酸缩水甘油酯发生反应。
实验组7
实验组7与实验组1不同的是,反应时间为1小时。
由此所获得的改性发泡剂发气量为49ml/g,分解气体中氨气含量达到7.3%。说明当反应时间过短时,反应发生不完全,部分ac没有与脂肪酸缩水甘油酯发生反应。
实施例2
实施例2提供了微发泡聚丙烯材料的制备方法。
实验组a
实验组a提供了一种微发泡聚丙烯材料,其包括:
聚丙烯树脂,所述聚丙烯树脂的熔融指数为38g/10min;
实验组1制备得到的改性发泡剂,改性发泡剂与聚丙烯树脂的质量百分比为1%;
成核剂(3,4-二甲基亚苄基山梨醇),3,4-二甲基亚苄基山梨醇与聚丙烯树脂的质量百分比为16%;
抗氧剂(dltp),dltp与聚丙烯树脂的质量百分比为0.2%;
润滑剂(硬脂酸钙),硬脂酸钙与聚丙烯树脂的质量百分比为0.8%。
通过下述方法制备上述微发泡聚丙烯材料:
在聚丙烯树脂中添加改性发泡剂,成核剂,抗氧剂,润滑剂,混合均匀,采用双螺杆挤出机熔融挤出造粒,螺杆挤出加工温度为180℃,螺杆转速为200rpm。制备的颗粒料注塑标准样条,成型温度230℃,由此获得上述微发泡聚丙烯注塑件。
将所制备得到的微发泡聚丙烯注塑件淬断,截面置于电镜下拍照,其结果如图1所示。从图1可以看出,所获得的改性发泡剂泡孔尺寸均一,分布均匀。
将所制备获得的微发泡聚丙烯材料进行测试,结果如表1所示。
实验组b
实验组b提供了一种微发泡聚丙烯材料,其包括:
聚丙烯树脂,所述聚丙烯树脂的熔融指数为38g/10min;
实验组2制备得到的改性发泡剂,改性发泡剂与聚丙烯树脂的质量百分比为3%;
成核剂(甲撑双(4,6-二叔丁基苯基)磷酸钠),甲撑双(4,6-二叔丁基苯基)磷酸钠与聚丙烯树脂的质量百分比为1%;
抗氧剂(1010),1010与聚丙烯树脂的质量百分比为0.5%;
润滑剂(聚乙烯蜡),聚乙烯蜡与聚丙烯树脂的质量百分比为0.5%。
通过下述方法制备上述微发泡聚丙烯材料:
在聚丙烯树脂中添加改性发泡剂,成核剂,抗氧剂,润滑剂,混合均匀,采用双螺杆挤出机熔融挤出造粒,螺杆挤出加工温度为190℃,螺杆转速为200rpm。制备的颗粒料注塑标准样条,成型温度230℃,由此获得上述微发泡聚丙烯注塑件。
将所制备获得的微发泡聚丙烯材料进行测试,结果如表1所示。
实验组c
实验组c提供了一种微发泡聚丙烯材料,其包括:
聚丙烯树脂,所述聚丙烯树脂的熔融指数为38g/10min;
实验组2制备得到的改性发泡剂,改性发泡剂与聚丙烯树脂的质量百分比为5%;
成核剂(滑石粉),滑石粉与聚丙烯树脂的质量百分比为30%;
抗氧剂(168),168与聚丙烯树脂的质量百分比为1%;
润滑剂(硬脂酸锌),硬脂酸锌与聚丙烯树脂的质量百分比为1%。
通过下述方法制备上述微发泡聚丙烯材料:
在聚丙烯树脂中添加改性发泡剂,成核剂,抗氧剂,润滑剂,混合均匀,采用双螺杆挤出机熔融挤出造粒,螺杆挤出加工温度为200℃,螺杆转速为200rpm。制备的颗粒料注塑标准样条,成型温度230℃,由此获得上述微发泡聚丙烯注塑件。
将所制备获得的微发泡聚丙烯材料进行测试,结果如表1所示。
实验组d
实验组d与实验组a不同的是,实验组d中所用到的改性发泡剂为实施例1中实验组3的改性发泡剂。
将所制备获得的微发泡聚丙烯材料进行测试,结果如表1所示。其气味测试结果较实验组a明显较差,说明利用实验组3的改性发泡剂作为发泡剂制备微发泡聚丙烯材料,所生成的氨气量较多。
实验组e
实验组e与实验组c不同的是,实验组e用到的改性发泡剂的量是聚丙烯树脂含量的10%。
将所制备获得的微发泡聚丙烯材料进行测试,结果如表1所示。其密度较实验组c并没有明显的下降,说明过量的发泡剂并不能进一步提高发泡倍率。由此为了节约成本和用量,可以适当降低改性发泡剂的用量,而不会产生不良影响。
实验组f
实验组f与实验组a不同的是,将改性发泡剂替换为摩尔比1:1.5的偶氮二甲酰胺和油酸缩水甘油酯混合物。
实验组f中仅仅将偶氮二甲酰胺和油酸缩水甘油酯进行物理混合,然后将其作为改性发泡剂制备微发泡聚丙烯材料,将所制备获得的微发泡聚丙烯材料进行测试,其结果如表1所示。其气味测试结果较实验组a大幅降低,结果较差,说明未改性的发泡剂生成的氨气量较多。
表1
从表1给出的结果不难看出,实验组a、实验组b、实验组c、实验组e等均获得了较好的微发泡聚丙烯材料。
尽管上面已经示出和描述了本发明的实施例,可以理解的是,上述实施例是示例性的,不能理解为对本发明的限制,本领域的普通技术人员在本发明的范围内可以对上述实施例进行变化、修改、替换和变型。
-
 0访客 来自[中国] 2023年05月13日 18:49联系我需要ac发泡剂改性技术15931192593
0访客 来自[中国] 2023年05月13日 18:49联系我需要ac发泡剂改性技术15931192593