纳米羟基磷灰石包覆的中空聚合物微球及其制备方法和应用与流程
本发明属于无机与高分子杂化材料领域,特别涉及一种纳米羟基磷灰石包覆的中空聚合物微球及其制备方法和应用。
背景技术:
中空聚合物微球由于内部具有空腔,因而表现出低密度、可以容纳客体分子等特点,在涂料、造纸、催化、分离、生物医药等领域有广阔的应用前景。
目前用于制备中空聚合物微球的方法主要有:模版法、自组装法、乳液法。模版法是在模版粒子的表面形成聚合物壳,再将模版粒子移除,形成中空聚合物壳。该法由于涉及制备并移除模版材料的过程,因而步骤繁琐,并且模版移除过程可能会对中空微球的结构产生很大的影响。自组装法是利用两亲嵌段共聚物在选择性溶剂中自组装形成胶束后通过聚合物间的交联作用将壳层固定从而得到中空聚合物微球。该法对聚合物结构的要求较高,须能形成稳定胶束并可进行交联反应。乳液法是制备中空聚合物微球最常用的方法,其过程相对简单、容易操作。但传统的乳化方法需要使用有机乳化剂,存在残留量高、具有毒性和环境污染等问题,而pickering乳化剂使用固体颗粒稳定油水体系,从而避免了上述问题。并且利用pickering乳液方法制备的聚合物微球表面覆盖有一层无机纳米粒子,可利用此将微球进一步功能化。
聚己内酯(poly(ε-caprolactone),pcl)具有良好的生物相容性和生物可降解性,被广泛用于生物医学工程领域。但是由于其疏水性较强,不利于细胞的粘附生长,因而常通过对其进行适当亲水改性的方式从而提高其细胞粘附性。
羟丙基纤维素(hydroxypropylcellulose,hpc)作为一种天然高分子,具有很好的生物相容性,并且hpc与其它聚合物具有良好的协同性和相容性。
目前利用pickering乳液法制备的聚合物微球主要是以聚苯乙烯实心微球为主。聚苯乙烯由于其生物相容性较差在生物医学领域的应用受到限制。到目前为止,尚未有纳米羟基磷灰石包覆的中空聚合物微球的相关报道。
技术实现要素:
本发明的首要目的在于克服现有技术的缺点与不足,提供一种纳米羟基磷灰石包覆的中空聚合物微球的制备方法。
本发明的另一目的在于提供所述方法制备得到的纳米羟基磷灰石包覆的中空聚合物微球。
本发明的再一目的在于提供所述纳米羟基磷灰石包覆的中空聚合物微球的应用。
本发明的目的通过下述技术方案实现:一种纳米羟基磷灰石包覆的中空聚合物微球的制备方法,包括如下步骤:
(1)将纳米羟基磷灰石(hap)分散到水,得到纳米羟基磷灰石分散液(作为水相);
(2)将羟丙基纤维素接枝己内酯共聚物(hpc-g-pcl)溶于有机溶剂中,得到羟丙基纤维素接枝己内酯共聚物溶液(作为油相);
(3)将步骤(1)中得到的纳米羟基磷灰石分散液加入到步骤(2)中得到的羟丙基纤维素接枝己内酯共聚物溶液中,震荡混合均匀,然后在常温下静置挥发,干燥,得到纳米羟基磷灰石包覆的中空聚合物微球(该微球具有中空结构,并且纳米羟基磷灰石包覆于微球表面)。
步骤(1)中所述的纳米羟基磷灰石(hap)为干燥后的纳米羟基磷灰石;优选为在37℃条件下真空干燥后的纳米羟基磷灰石。
步骤(1)中所述的纳米羟基磷灰石分散液的浓度为重量百分比0.1%~0.5%;优选为重量百分比0.1%。
步骤(2)中所述的羟丙基纤维素接枝己内酯共聚物(hpc-g-pcl)的反应方程式可表示为:
其中:
m:1~500;m′:1~500;n:1~500。
所述的羟丙基纤维素接枝己内酯共聚物(hpc-g-pcl)优选为通过如下方法制备得到:
将羟丙基纤维素(hpc)加入到ε-cl(ε-己内酯)中,待羟丙基纤维素溶解后升温至95~120℃,加入辛酸亚锡作为催化剂,在保护性气体进行反应,待反应结束后将所得产物溶解在四氢呋喃(thf)中,并加入正己烷使得生成的产物沉淀,重复三次,收集沉淀,干燥,得到羟丙基纤维素接枝己内酯共聚物(hpc-g-pcl)。
所述的羟丙基纤维素(hpc)与ε-cl(ε-己内酯)的质量比为1:8~12;优选1:10。
所述的升温优选为升温至105℃。
所述的辛酸亚锡的添加量为按每克(g)羟丙基纤维素配比0.4~0.6辛酸亚锡计算;优选按每克(g)羟丙基纤维素配比0.5辛酸亚锡计算。
所述的保护性气体优选为氮气。
所述的反应的时间为20~48小时;优选为24小时。
所述的四氢呋喃的用量为按每克(g)ε-cl(ε-己内酯)配比10~20ml四氢呋喃(thf)计算。
所述的己烷与四氢呋喃的体积比为1:1。
步骤(2)中所述的有机溶剂为易挥发有机溶剂;优选为二氯甲烷。
步骤(2)中所述的羟丙基纤维素接枝己内酯共聚物溶液的浓度为重量百分比0.2%~0.5%;优选为重量百分比0.3%。
步骤(3)中所述的羟丙基纤维素接枝己内酯共聚物溶液与纳米羟基磷灰石分散液的体积比(油水比)为1:3~6。
步骤(3)中所述的静置的时间为1~3天。
步骤(3)中所述的干燥为在37℃、真空条件下进行干燥。
一种纳米羟基磷灰石包覆的中空聚合物微球,通过上述任一项所述的方法制备得到。
所述的纳米羟基磷灰石包覆的中空聚合物微球在制备载药微球中的应用。
一种具有缓释作用的载药微球的制备方法,包括如下步骤:
(i)将纳米羟基磷灰石(hap)分散到水,得到纳米羟基磷灰石分散液(作为水相);
(ii)将羟丙基纤维素接枝己内酯共聚物(hpc-g-pcl)和油溶性药物溶于有机溶剂中,得到混合溶液(作为油相);
(iii)将步骤(i)中得到的纳米羟基磷灰石分散液加入到步骤(ii)中得到的混合溶液中,震荡混合均匀,然后在常温下静置挥发,干燥,得到纳米羟基磷灰石包覆的中空聚合物微球。
步骤(i)中所述的纳米羟基磷灰石分散液的浓度为重量百分比0.1%~0.3%;优选为重量百分比0.1%。
步骤(ii)中所述的羟丙基纤维素接枝己内酯共聚物(hpc-g-pcl)优选为通过如下方法制备得到:
将羟丙基纤维素(hpc)加入到ε-cl(ε-己内酯)中,待羟丙基纤维素溶解后升温至95~120℃,加入辛酸亚锡作为催化剂,在保护性气体进行反应,待反应结束后将所得产物溶解在四氢呋喃(thf)中,并加入正己烷使得生成的产物沉淀,重复三次,收集沉淀,干燥,得到羟丙基纤维素接枝己内酯共聚物(hpc-g-pcl)。
所述的羟丙基纤维素(hpc)与ε-cl(ε-己内酯)的质量比为1:8~12;优选1:10。
所述的升温优选为升温至105℃。
所述的辛酸亚锡的添加量为按每克(g)羟丙基纤维素配比0.4~0.6辛酸亚锡计算;优选按每克(g)羟丙基纤维素配比0.5辛酸亚锡计算。
所述的保护性气体优选为氮气。
所述的反应的时间为20~48小时;优选为24小时。
所述的四氢呋喃的用量为按每克(g)ε-cl(ε-己内酯)配比10~20ml四氢呋喃(thf)计算。
所述的己烷与四氢呋喃的体积比为1:1。
步骤(ii)中所述的有机溶剂为易挥发有机溶剂;优选为二氯甲烷。
步骤(ii)中所述的混合溶液中羟丙基纤维素接枝己内酯共聚物溶液的浓度为重量百分比0.2%~0.5%(优选为0.3%);油溶性药物的浓度为重量百分比0.1~0.3%。
步骤(ii)中所述的油溶性药物为吲哚美辛,喜树碱和紫杉醇中的至少一种;优选为吲哚美辛。
步骤(iii)中所述的混合溶液与纳米羟基磷灰石分散液的体积比(油水比)为1:3~6。
步骤(iii)中所述的静置的时间为1~3天。
步骤(iii)中所述的干燥为在37℃、真空条件下进行干燥。
一种具有缓释作用的载药微球,通过上述任一项所述的方法制备得到。
本发明相对于现有技术具有如下的优点及效果:
(1)本发明制备纳米羟基磷灰石包覆的中空聚合物微球所用的原料为纳米羟基磷灰石、羟丙基纤维素、聚己内酯,均为具有良好生物相容性的物质,对生物体无毒害,对环境无污染。
(2)羟丙基纤维素具有活性基团羟基,因而本发明利用羟丙基纤维素分子结构中的-oh作为反应官能团,使其进一步接枝改性反应,即可得到新的改性材料。
(3)本发明利用羟丙基纤维素上的羟基作为引发剂,使己内酯在羟丙基纤维素分子上发生开环聚合反应(己内酯通过开环聚合反应接枝在羟丙基纤维素分子上),制备羟丙基纤维素接枝己内酯共聚物(hpc-g-pcl),并控制原料的投料比,使得聚合物具有一定的两亲性,同时又可以达到改善聚己内酯(pcl)亲水性的目的,从而更利于细胞的粘附生长。
(4)本发明利用纳米羟基磷灰石作为颗粒乳化剂,稳定ch2cl2/h2o油水体系,形成pickering乳液,制备油相ch2cl2中溶有hpc-g-pcl的o/w乳液,再通过将油相溶剂ch2cl2挥发,从而得到表面覆盖hap的hpc-g-pcl中空微球。
(5)本发明直接使用具有一定两亲性的聚合物溶液作为油相,随着油相溶剂的挥发,聚合物由于具有两亲性,会自发地向油水界面移动,当油相溶剂完全挥发,即得到以聚合物为壳且表面覆盖有无机粒子的中空微球。该制备过程简单易行,无需再进行除核和去除乳化剂过程。
(6)本发明制备的纳米羟基磷灰石包覆的中空聚合物微球不仅其本身具有良好的生物相容性和生物活性,而且其具有较大的空腔,可以对油溶性药物等实现包覆并达到缓释效果,其在生物医药领域具有潜在应用价值。
(7)本发明的纳米羟基磷灰石包覆的中空聚合物微球,不但其制备条件温和、操作简便、易于实施,而且具有较大空腔,对油溶性药物等具有较强的包封能力。
附图说明
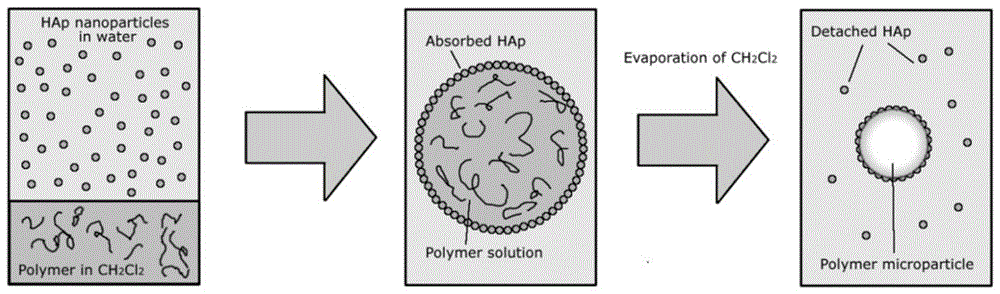
图1是本发明中hap包覆的中空聚合物微球的制备过程图。
图2是本发明所形成乳液的光学显微镜表征图;其中,a与b为同一视野下于相同放大倍数相隔半分钟拍摄结果。
图3是本发明所制备的纳米羟基磷灰石包覆的中空聚合物微球的扫描电镜(sem)表征图;其中,a~f分别为在不同视野下观察到的微球形貌。
图4是不同油水比下所制备微球的形态表征及粒径统计图;其中,a为油水比1:3时所制备的微球,平均直径66.8μm;b为油水比1:4时所制备的微球,平均直径60.7μm;c为油水比1:5时所制备的微球,平均直径35.9μm;d为油水比1:6时所制备的微球,平均直径44.0μm。
图5是本发明制备的载药微球的药物释放曲线图(图中:a为吲哚美辛与hpc-g-pcl质量比为1:3时所制备的载药微球的释放曲线;b为吲哚美辛与hpc-g-pcl质量比为2:3时所制备的载药微球的释放曲线;c为吲哚美辛与hpc-g-pcl质量比为1:1时所制备的载药微球的释放曲线)。
具体实施方式
下面结合实施例对本发明作进一步详细的描述,但本发明的实施方式不限于此。除非特别说明,本发明采用的试剂、方法和设备为本技术领域常规试剂、方法和设备。除非特别说明,本发明所用试剂和原材料均可通过市售获得。
实施例1hap包覆的hpc-g-pcl微球的制备
(1)羟丙基纤维素接枝己内酯共聚物(hpc-g-pcl)的合成
将ε-cl(ε-己内酯,阿拉丁试剂公司)与羟丙基纤维素(hydroxypropylcellulose,hpc)充分干燥(37℃真空干燥)后,称取1g液态的ε-cl,将0.1ghpc加入到ε-cl中,待hpc在ε-cl中溶解完全后将反应温度升至105℃,加入50μl辛酸亚锡作为催化剂,氮气保护下反应24小时,将所得产物溶解在10~20ml四氢呋喃(thf)中,加入与thf等体积的正己烷使得生成的hpc-g-pcl沉淀,重复三次后将收集到的沉淀物干燥。根据称重法对产物接枝率进行计算,得接枝率gy=790%,其中计算公式如式1-1所示:
gy=(w2-w1)×100/w1(1-1)
式中:w2为接枝后产物重量,w1为接枝前hpc重量。
(2)hap包覆的hpc-g-pcl微球的制备(图1)
①将0.1g纳米羟基磷灰石(hydroxylapatite,hap)分散于100ml水中,制备得到0.1wt%的hap分散液,作为水相;
②将0.3ghpc-g-pcl溶于10ml二氯甲烷中,得到3wt%的hpc-g-pcl的二氯甲烷溶液,作为油相;
③取1ml油相溶液,加入5ml水相分散液(油水比1:5),震荡得到乳液,于常温静置挥发(静置1~3天),37℃真空干燥,得到纳米羟基磷灰石包覆的中空聚合物微球。所制备的微球具有中空结构,壳层为羟丙基纤维素接枝聚己内酯聚合物形成,且表面包覆有纳米羟基磷灰石。
混合所形成乳液的光学显微镜图如图2所示,图2a与2b为同一视野下于相同放大倍数相隔半分钟拍摄结果,可以看出二氯甲烷会从形成的乳液滴中挥发。
制备的纳米羟基磷灰石包覆的中空聚合物微球的扫描电镜(sem)图如图3所示,图中表明所制备的微球为中空结构,并且纳米羟基磷灰石覆盖于微球表面。
实施例2吲哚美辛(indomethacin)载药微球的制备
(1)羟丙基纤维素接枝己内酯共聚物(hpc-g-pcl)的合成
同实施例1步骤(1)。
(2)hap包覆的hpc-g-pcl载药微球的制备
①将0.1g纳米羟基磷灰石(hydroxylapatite,hap)分散于100ml水中,制备得到0.1wt%的hap分散液,作为水相;
②将0.3ghpc-g-pcl及0.1g油溶性药物吲哚美辛(indomethacin)溶于10ml二氯甲烷中,得到3wt%(按hpc-g-pcl计算)的hpc-g-pcl与吲哚美辛的二氯甲烷混合溶液,作为油相;
③取1ml油相溶液,加入5ml水相分散液(油水比1:5),震荡得到乳液,于常温静置挥发(静置1-3天),37℃真空干燥,得到吲哚美辛载药微球。所制备的载药微球的空腔内部载有油溶性药物,羟丙基纤维素接枝聚己内酯聚合物为壳层,并且表面包覆有纳米羟基磷灰石。
实施例3
按照实施例2的方法,不同之处在于:步骤(2)③中油水比替换为1:3(取1ml油相溶液,加入3ml水相分散液),1:4(取1ml油相溶液,加入4ml水相分散液),1:6(取1ml油相溶液,加入6ml水相分散液),分别制备得到吲哚美辛载药微球。
将不同油水比下所制备吲哚美辛载药微球在扫描电镜(sem)下观察并统计其粒径,结果如图4所示:4a为油水比1:3时所制备的微球,平均直径66.8μm;4b为油水比1:4时所制备的微球,平均直径60.7μm;4c为油水比1:5时所制备的微球,平均直径35.9μm;4d为油水比1:6时所制备的微球,平均直径44.0μm。
实施例4
(1)按照实施例2的方法,不同之处在于:步骤(2)②中吲哚美辛与hpc-g-pcl质量比为1:3(0.1g吲哚美辛,0.3ghpc-g-pcl),2:3(0.2g吲哚美辛,0.3ghpc-g-pcl),1:1(0.3g吲哚美辛,0.3ghpc-g-pcl),分别制备得到吲哚美辛载药微球。
(2)分别称取5mg吲哚美辛载药微球,于截留分子量3000的透析袋中,扎紧后置于释放液中,其中释放液为10ml含10%(v/v)乙醇的pbs缓冲液(0.01m,ph7.4),于37℃恒温水浴箱中震荡释放。每次取样时(0~48小时内定点取样),取出3ml,再加入3ml新鲜释放液。然后用紫外分光光度计在320nm处测量吸光度值,绘制药物释放曲线。
通过紫外测定所制备的载药微球对吲哚美辛的包封率达98%(包封率=实际包裹量/药物使用量),并且对药物的控制释放有明显效果,无明显爆释现象,释放过程可持续24h,其药物释放曲线如图5所示。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!