一种印乌碱砂炒炮制过程中的产物及其制备方法和用途与流程
本发明属于医药化学领域,具体涉及一种印乌碱砂炒炮制过程中的产物及其制备方法和用途。
背景技术:
毛茛科(ranunculaceae)乌头属aconituml.植物是一类重要的药用植物,主要治疗跌打损伤痛、风湿痛、腰腿痛等疾病。全世界有300余种,分布于北半球温带地区;我国有200多种,主要分布于四川西部、云南北部、西藏东部的高山地带,以及东北、西北和内蒙古地区,其中有40种可供药用,如川乌a.carmichaelii/radix、北乌头a.kusnezoffiireichb.、黄花乌头a.coreanum(h.lév.)rapaics、短柄乌头a.brachypodumdiels、铁棒锤a.pendulumbusch、康定乌头aconitumtatsienensefinet&gagnep.等。但乌头类植物含有二萜生物碱成分,具有较强的毒性,中毒表现为唇、舌、四肢麻木、心悸、心率减慢或心动过速、肌肉强直、呼吸痉挛、窒息而危及生命。因此,需要炮制后使用。
目前,中医对于乌头类药材的炮制主要采用水煮法(加水煮沸4~6小时)及蒸制法(蒸6~8小时),其炮制减毒原理为:有毒生物碱通过酯键水解反应,由双酯型生物碱依次转化为毒性较低的单酯型和醇胺型生物碱,如乌头碱转化为苯甲酰乌头原碱和乌头原碱。
与中医采用水煮法和蒸制法不同,藏、羌医通常采用砂炒法炮制乌头类药材,仅需炒制5~10分钟即可达到减毒的目的。但砂炒法的炮制原理尚未完全阐明,砂炒过程中毒性生物碱的结构如何转化,转化产物与原型生物碱相比,毒性是否降低尚未明确。并且由于砂炒法的炮制原理尚未阐明,导致砂炒品的质量仍以药材炒至“外皮焦黄,断面发黄”等主观经验来判断,缺乏定量化的质控指标。
另外有研究表明,有些二萜生物碱虽然结构相近,但毒性和功效却不相同,例如有的能够引起心律失常,具有心脏毒性,有的却有治疗心律失常的作用。因此,在比较砂炒产物与原型生物碱毒性的基础上,有必要对砂炒产物是否具有抗心律失常作用进行研究。
技术实现要素:
本发明的目的是提供一种印乌碱砂炒炮制过程中的产物及其制备方法和用途。
本发明提供了式i所示化合物、或其盐:
其中,
r1选自氢、c1~c8烷基、c1~c8烷氧基、卤素、-oc(o)r2;
r2选自c1~c8烷基。
进一步地,
r1选自氢、c1~c3烷基、c1~c3烷氧基、-oc(o)r2;
r2选自c1~c6烷基。
进一步地,
r1选自-oc(o)r2;
r2选自c1~c3烷基。
进一步地,所述化合物为式ii所示化合物:
本发明还提供了一种前述化合物、或其盐的制备方法,它包括如下步骤:
(1)取印乌碱溶于溶剂中,蒸干溶剂,油浴,得印乌碱炮制产物;
(2)将印乌碱炮制产物进行硅胶柱层析,梯度洗脱后合并具有式ii所述化合物的洗脱液流分,得到组分c;
(3)将组分c过硅胶柱色谱,洗脱后,即得;
步骤(1)中,所述油浴为140℃油浴20~40min,或160℃油浴10~40min,或180℃油浴10~20min。
进一步地,
步骤(1)中,所述溶剂为二氯甲烷;
和/或,步骤(1)中,所述印乌碱和溶剂的质量体积比为(5~10):1(mg/ml);
和/或,步骤(1)中,所述蒸干溶剂为用旋转薄膜蒸发仪在40℃条件下蒸干溶剂;
和/或,步骤(1)中,所述油浴为160℃下油浴30min;
和/或,步骤(2)中,所述硅胶柱为200-300目的硅胶柱;
和/或,步骤(2)中,所述梯度洗脱的洗脱剂为石油醚-丙酮-三乙胺混合溶液,石油醚-丙酮-三乙胺的体积比依次为8:1:0.01,6:1:0.01,3:1:0.01;
和/或,步骤(2)中,所述梯度洗脱中,每个梯度洗脱液的用量与印乌碱炮制产物的体积质量比为(10~20):3(ml/mg);
和/或,步骤(3)中,所述硅胶柱为200-300目的硅胶柱;
和/或,步骤(3)中,所述洗脱的洗脱剂为石油醚-丙酮-三乙胺混合溶液,石油醚-丙酮-三乙胺的体积比为3:1:0.01。
进一步地,
步骤(1)中,所述印乌碱和溶剂的质量体积比为5.2:1(mg/ml);
和/或,步骤(2)中,所述梯度洗脱中,每个梯度洗脱液的用量与印乌碱炮制产物的体积质量比为10:3(ml/mg)。
本发明还提供了前述的化合物、或其盐在制备治疗心律失常的药物中的用途。
本发明还提供了一种药物,它是由前述的化合物、或其盐为活性成分,加上药学上接受的辅助性成分或辅料制备而成的制剂。
本发明中,碳氢基团中碳原子含量的最小值和最大值通过前缀表示,例如,前缀ca~cb烷基表明任何含“a”至“b”个碳原子的烷基,因此,例如,c1~c6烷基是指包含1~6个碳原子的烷基。又例如,前缀ca~cb烷氧基表明任何含“a”至“b”个碳原子的烷氧基,因此,例如c1~c6烷氧基是指包含1~6个碳原子的烷氧基。
本发明中,-oc(o)r2的结构式为
本发明从印乌碱炮制品中分离得到的化合物δ15,16-16-demethoxyindaconitine是一种新化合物,与印乌碱相比,该化合物安全性高,并且具有优异的治疗心律失常作用,可用于制备治疗心律失常的药物,可以大大降低含有印乌碱的乌头类药物的毒性,具有良好的应用前景。
显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离本发明上述基本技术思想前提下,还可以做出其它多种形式的修改、替换或变更。
以下通过实施例形式的具体实施方式,对本发明的上述内容再作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。
附图说明
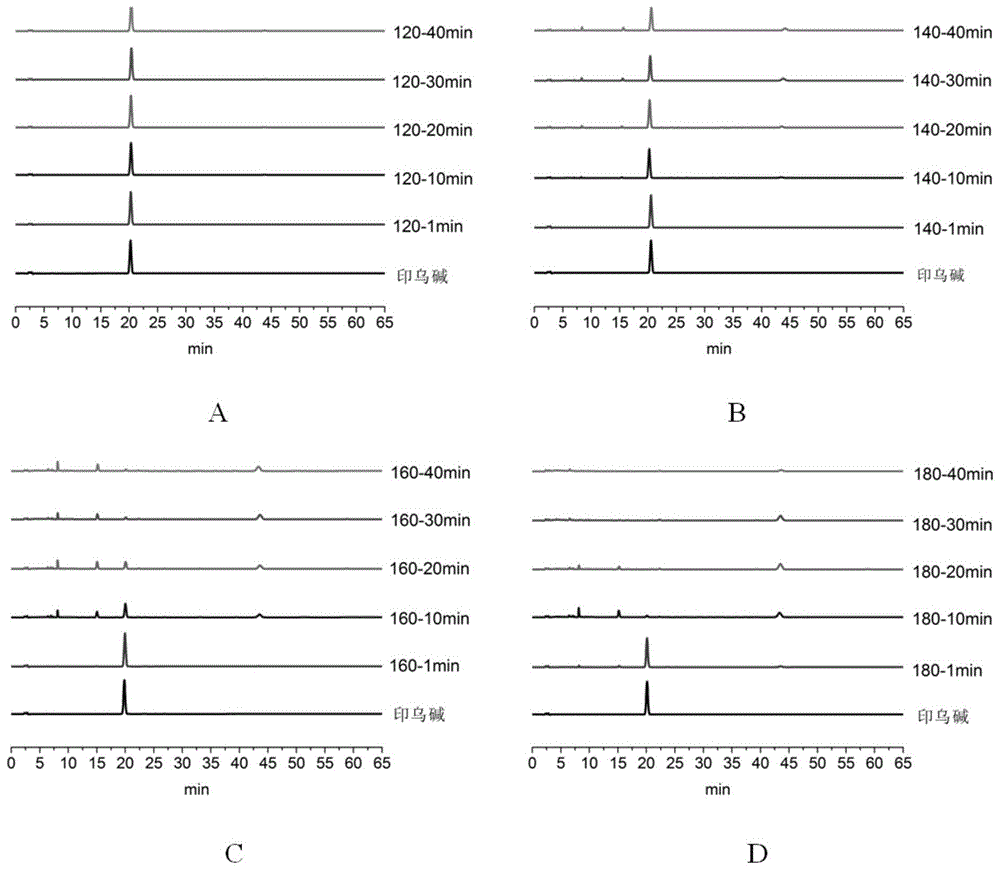
图1为印乌碱在不同炮制温度和时间条件下的色谱图;a:120℃;b:140℃;c:160℃;d:180℃。
图2为印乌碱在不同温度、时间条件下含量变化趋势图。
图3为化合物2(δ15,16-16-demethoxyindaconitine)的1h-1hcosy
图4为sd大鼠特征心电图;a为正常心电图;b、c、d分别为室性早搏、室性心动过速、室颤心电图。
图5为δ15,16-16-demethoxyindaconitine对sd大鼠vpb潜伏期的影响;与模型对照组比较:*p<0.05;与普罗帕酮组比较:#p<0.05;与利多卡因组相比:+p<0.05。
图6为δ15,16-16-demethoxyindaconitine不同剂量组vt发生率;与0.05mg/kg剂量组比较:*p<0.05;与0.10mg/kg剂量组比较:#p<0.05。
图7为各受试药物组心律失常完全抑制率;与≤0.20mg/kg剂量组比较:*p<0.05。
具体实施方式
1、仪器与试药
agilent1260型高效液相色谱仪,agilentchemastation色谱工作站;bl-420f多功能生理记录仪(成都泰盟软件有限公司);cpa2250电子分析天平(德国sartorius公司);ulup-i-10t优普超纯水机(成都超纯科技有限公司);hh-sj集热式磁力搅拌器(金坛市城东新瑞仪器厂);xt-4显微熔点仪(上海精密科学仪器有限公司);perkin-elmer341polarimeter旋光仪;brukeravance600spectrometer核磁共振仪,tms为内标;brukermicro-tof-q-ⅱmassspectrometer质谱仪;薄层色谱硅胶g和柱色谱硅胶(200-300目)(青岛海洋化工厂);印乌碱(t-043-161216,纯度>98%,陕西昊辰生物科技有限公司);乌头碱(wtz10013,纯度99%,陕西昊辰生物科技有限公司);盐酸普罗帕酮(101190-201702,纯度99.8%,中国食品药品检定研究院);盐酸利多卡因(100341-201403,纯度93.4%,中国食品药品检定研究院);乌拉坦(2017090501,成都市科龙化工试剂厂);生理盐水(l218090910,四川科伦药业股份有限公司);色谱所用石油醚沸点60-90℃,展开比例为v/v;显色剂为改良碘化铋钾试剂;乙腈为色谱纯(美国fisher公司);水为超纯水;其余试剂均为分析纯。
2、实验动物
spf级健康成年sd大鼠,雌雄兼用,体重(200±20)g均由成都达硕实验动物有限公司提供,生产许可证号:scxk(川)-2015-030。
实施例1、印乌碱炮制工艺参数的筛选
1、色谱条件
色谱柱:cosmosilpak5c18-ms-ⅱ(4.6mm×250mm,5μm);流动相:乙腈-0.03mol/l碳酸氢铵溶液(浓氨水调ph9.5)(43:57)等度洗脱;柱温:35℃;流速:1.0ml/min;检测波长:230nm。
2、对照品溶液的制备
取印乌碱46mg,精密称定,置100ml量瓶中,加二氯甲烷制成浓度为0.4626mg/ml的反应储备液。取4ml该反应储备液,置10ml量瓶中,挥干溶剂,加0.1%盐酸-甲醇定容,制成0.185mg/ml的对照品溶液,摇匀,过微孔滤膜(0.45μm),取续滤液,即得对照品溶液。
3、炮制品供试溶液的制备
取圆底烧瓶20个,分为120℃、140℃、160℃、180℃四个温度组,每个温度下设置五个时间点(1、10、20、30、40min),分别加入“2、对照品溶液的制备”中的反应储备液4ml,挥干溶剂后,按照设置的温度、时间进行加热,反应结束后,放冷,加0.1%盐酸-甲醇溶解,并转移至10ml量瓶中,加0.1%盐酸-甲醇定容至刻度,作为炮制品供试溶液,摇匀,过微孔滤膜(0.45μm),取续滤液,即得。
4、方法学考察
4.1线性关系、检测限和定量限考察
取印乌碱适量,加0.1%盐酸-甲醇制成浓度为2mg/ml的储备液,精密吸取该储备液0.025、0.1、0.2、0.4、0.8、1.0、2.0、3.0ml置10ml量瓶中,加0.1%盐酸-甲醇定容至刻度,制成浓度为5μg/ml、20μg/ml、40μg/ml、80μg/ml、160μg/ml、200μg/ml、400μg/ml、600μg/ml的溶液,摇匀,过微孔滤膜(0.45μm),取续滤液,即得不同浓度的印乌碱溶液。
分别精密吸取“4.1”制备的不同浓度的印乌碱溶液10μl注入液相色谱仪,按“1、色谱条件”的参数,测定峰面积,并以进样量为横坐标(x),以峰面积为纵坐标(y),进行线性回归,得回归方程为y=11.913x+1.3739,r=0.9992,表明在5~600μg/ml范围内线性关系良好。
根据3倍和10倍信噪比所对应的印乌碱的浓度,确定方法的检测限(lod)为1.075μg/ml;定量限(loq)为3.44μg/ml。
4.2精密度试验
精密吸取“2”制备的印乌碱对照品溶液10μl,按“1、色谱条件”,连续进样6次,得印乌碱峰面积的rsd为1.06%,表明精密度良好。
4.3稳定性试验
精密吸取“2”制备的印乌碱对照品溶液,放置0、1、2、4、8、12h后,按“1、色谱条件”进行测定,进样量10μl。测得印乌碱峰面积的rsd为1.30%,表明对照品溶液在12h内稳定性良好。
5、试验结果
分别吸取对照品与炮制品供试溶液注入高效液相色谱仪,按“1、色谱条件”进行测定,得到不同温度、时间条件下色谱图。通过测定印乌碱峰面积并比较不同加热温度、时间的供试品中有无新的色谱峰出现,能够直观地反映出印乌碱在不同温度、时间条件下的转化过程,结果见图1、图2、表1。
表1、不同炮制品中印乌碱含量测定结果(μg/ml)
炮制前印乌碱含量为185.04μg/ml,在160℃加热炮制10min后,含量降为74.82μg/ml,炮制20min后含量降为38.47μg/ml,180℃炮制10min后,含量降为8.65μg/ml,20min时,含量降为0μg/ml。观察图1-b、1-c、图1-d发现,140℃-20~40min、160℃-10~40min及180℃-10~20min色谱图中,在保留时间8min、15min和43min左右处,出现了三个新的色谱峰,提示在这三个温度及相应时间条件下,印乌碱有三个转化产物。
实施例2、印乌碱炮制转化成分的分离和结构鉴定
1、柱色谱分离样品的制备
取印乌碱520mg,置250ml圆底烧瓶中,加二氯甲烷100ml溶解,用旋转薄膜蒸发仪在40℃条件下蒸干溶剂,使印乌碱均匀地附着在圆底烧瓶内壁,并于160℃下油浴30min,取出,放凉,得到印乌碱炮制产物(450mg),供上柱用。
2、分离纯化
将印乌碱炮制产物进行硅胶柱层析(200-300目,120g),石油醚-丙酮-三乙胺(8:1:0.01,6:1:0.01,3:1:0.01)梯度洗脱,每个梯度1.5l,薄层板检视,合并含有化合物2的流分,得到组分c。组分c(100mg)进一步采用硅胶柱色谱(200-300目,100g),洗脱剂为石油醚-丙酮-三乙胺(3:1:0.01),得到化合物2(50mg)。
化合物2结构式的解析过程如下:
化合物2:白色晶体,mp:93~95℃,
化合物2与印乌碱的nmr图谱相似,但从13c-nmr中可以看出:(1)化合物2的c-16位缺少一个-och3;(2)印乌碱的c-15、c-16的化学位移分别为39.4和82.8,而化合物2的c-15、c-16的化学位移分别向低场迁移到125.5和137.1。因此,在1h-nmr中,δh6.54(1h,d,j=10.3hz)和δh6.06(1h,d,j=10.3hz)分别归属为h-15和h-16,表明c-15和c-16之间形成了双键,hmbc谱中,c-13与h-15,c-8与h-16远程相关,也证实c-15与c-16之间存在双键结构(图3),根据1h-nmr,13c-nmr及2d-nmr(hmbc,1h-1hcosy,noesy)数据(表2),可以确定化合物2的结构为δ15,16-16-demethoxyindaconitine(15,16-didehydro-1α,6α,-dimethoxy-4β-(methoxymethyl)-20-ethyl-aconitane-3α,13β,14α-triol14-benzoate),化合物2为新化合物。
化合物2结构式如下所示:
表2、化合物2(δ15,16-16-demethoxyindaconitine)的nmr数据
以下通过具体的试验例证明本发明的有益效果。
试验例1、印乌碱及其砂炒产物δ15,16-16-demethoxyindaconitine的心脏毒性试验
1、试验方法
取sd大鼠20只,雌雄各半,随机分为2组(n=10),即印乌碱组和δ15,16-16-demethoxyindaconitine(实施例2制备的化合物2)组,通过预实验,确定0.06mg/kg的印乌碱即可引起大鼠室性心律失常,如室性早搏(ventricularprematurebeat,vpb)和室性心动过速(ventriculartachyrhythmia,vt);通过观察相同剂量下其砂炒产物δ15,16-16-demethoxyindaconitine是否引起心律失常,可直观反映出经过炮制后,印乌碱结构上的改变是否引起其毒性的降低。
数据采用spss统计软件处理,vpb潜伏期实验结果以
2、试验结果
在0.06mg/kg剂量下,印乌碱可引起大鼠室性心律失常(包括vpb和vt),胸部伴随出现规律的抽搐现象;在相同剂量下,δ15,16-16-demethoxyindaconitine组大鼠未出现心律失常反应(包括心电图变化和大鼠的行为反应)。因此,与印乌碱相比,其砂炒产物δ15,16-16-demethoxyindaconitine的毒性显著降低,如表3、图4所示。
表3、印乌碱及砂炒产物的毒性比较
“—”表示在给药30min内,未出现vpb。
试验例2、δ15,16-16-demethoxyindaconitine的抗心律失常活性研究
1、试验方法
取90只sd大鼠,雌雄各半,随机分为9组(n=10),即溶剂对照组(溶剂的配制:4ml1%盐酸乙醇,加生理盐水定容至100ml)、模型对照组(生理盐水)、普罗帕酮组(剂量为3.20mg/kg,使用溶剂对照组的溶剂配制)、利多卡因组(剂量为5.00mg/kg,使用溶剂对照组的溶剂配制)及不同剂量δ15,16-16-demethoxyindaconitine组(剂量为0.05mg/kg、0.10mg/kg、0.20mg/kg、0.30mg/kg、0.40mg/kg,使用溶剂对照组的溶剂配制)。各组sd大鼠,乌拉坦1.2g/kg腹腔注射麻醉后,仰位固定,针型电极插入四肢皮下,静置观察并记录ⅱ导联心电图(electrocardiogram,ecg);20min后,δ15,16-16-demethoxyindaconitine各剂量组分别经股静脉快速推注不同剂量的δ15,16-16-demethoxyindaconitine,两个阳性药对照组分别注射相应剂量的普罗帕酮和利多卡因,模型对照组给等量生理盐水,溶剂对照组给对应剂量的溶剂。观察大鼠反应并记录ⅱ导联ecg;10min后,各组(除溶剂对照组外)再经股静脉快速推注乌头碱(0.03mg/kg),分别记录出现vpb、vt等心律失常现象的时间(即潜伏期)并观察静注完乌头碱30min内是否有未出现心律失常的情况发生。
数据采用spss统计软件处理,vpb潜伏期实验结果以
2、试验结果
(1)δ15,16-16-demethoxyindaconitine对乌头碱诱发大鼠vpb潜伏期的影响
vpb是心律失常发生初期的表现,与正常心电图相比,主要表现为qrs波提前出现,形态异常,t波与qrs主波方向相反,且p波消失。vpb潜伏期是指注射乌头碱后,至vpb出现的时间,该时间段越长,表明受试化合物的抗心律失常作用越强。
乌头碱致心律失常模型对照组中,sd大鼠vpb潜伏期为(97.7±23.7)s;普罗帕酮组(pro)为(142.3±17.5)s,利多卡因组(lid)为(180.9±51.0)s;与模型对照组相比,两种阳性药均有显著性差异(p<0.05)。
δ15,16-16-demethoxyindaconitine不同剂量组(0.05mg/kg、0.10mg/kg、0.20mg/kg、0.30mg/kg、0.40mg/kg)的vpb潜伏期分别为:(161.9±63.1)s,(224.2±101.6)s,(363.4±105.0)s,(896.5±239.3)s,(1211.0±389.1)s;与模型对照组相比,除了0.05mg/kg组外,其他剂量组均有显著性差异(p<0.05);与普罗帕酮及利多卡因组相比,0.20mg/kg、0.30mg/kg及0.40mg/kg组均有显著性差异(p<0.05),如图5所示。
(2)δ15,16-16-demethoxyindaconitine对大鼠vt发生率的影响
vt是vpb进一步发展的结果,其心电图特点为:连续出现3次或3次以上的室性期前收缩,qrs波群宽大畸形,无恒定的p波。因此,vt发生率可以评价受试药物是否能够控制心律失常的进一步发展。
为了探究δ15,16-16-demethoxyindaconitine不同剂量组出现心动过速的情况,将δ15,16-16-demethoxyindaconitine分为4个组:0.05mg/kg、0.10mg/kg、0.20mg/kg和0.30~0.40mg/kg组。卡方检验结果显示:这4个剂量组的vt发生率有差异(p<0.05),如表4所示;两两比较结果显示:与0.05mg/kg及0.10mg/kg组相比,0.30~0.40mg/kg剂量组的vt发生率显著降低(p<0.05),见表5~6、图6。
表4、δ15,16-16-demethoxyindaconitine不同剂量组vt发生率分析
注:p<0.05有统计学意义。
表5、0.05mg/kg与0.30~0.40mg/kg剂量组vt发生率两两比较分析
注:p<0.05有统计学意义。
表6、0.10mg/kg与0.30~0.40mg/kg剂量组vt发生率两两比较分析
注:p<0.05有统计学意义。
(3)δ15,16-16-demethoxyindaconitine对大鼠心律失常完全抑制率的影响
心律失常总发生率是指大鼠预先注射受试药物,并用乌头碱建立心律失常模型后,30min内出现心律失常的比例(包括早搏、心动过速或室颤)。心律失常完全抑制率=100%-心律失常总发生率,即30min内未出现心律失常的比例,可以评价受试药物是否能够完全拮抗乌头碱的致心律失常作用。
本研究文比较了δ15,16-16-demethoxyindaconitine不同剂量组(≤0.20mg/kg、0.30mg/kg、0.40mg/kg)及阳性药利多卡因(lid)对大鼠的心律失常完全抑制率,卡方检验结果显示:4个剂量组的心律失常完全抑制率有差异(p<0.05)(表7)。两两比较结果显示,与≤0.20mg/kg组相比,δ15,16-16-demethoxyindaconitine的0.40mg/kg剂量组以及利多卡因组的心律失常完全抑制率均显著升高(p<0.05);见表8、图7。
表7、心律失常完全抑制率分析
注:p<0.05有统计学意义。
表8、心律失常完全抑制率两两比较分析
注:p<0.05有统计学意义。
综上,本发明从印乌碱炮制品中分离得到的化合物δ15,16-16-demethoxyindaconitine是一种新化合物,与印乌碱相比,该化合物安全性高,并且具有优异的治疗心律失常作用,可用于制备治疗心律失常的药物,可以大大降低含有印乌碱的乌头类药物的毒性,具有良好的应用前景。
- 还没有人留言评论。精彩留言会获得点赞!