一种拉考沙胺的合成方法与流程
本发明涉及医药化学领域,具体涉及一种治疗癫痫和神经性疼痛的药物拉考沙胺的合成方法。
背景技术:
拉考沙胺(lacosamide)是一种新型n-甲基-d-天门冬氨酸(nmda)受体甘氨酸位点结合拮抗剂,属于新一类功能性氨基酸,适用于16岁及以上癫痫患者部分性发作的联合治疗。于2008年由美国fda和欧洲ema批准上市,可通过口服或静脉注射给药。癫痫,是神经科仅次于头痛的第二大常见病,已成为全球性公共卫生问题,其发病机理是大脑神经元突发性异常放电,导致短暂的大脑功能障碍的一种慢性疾病。据中国最新流行病学资料显示,国内癫痫的总体患病率为7.0‰,年发病率为28.8/10万,1年内有发作的活动性癫痫患病率为4.6%。据此估计中国约有900万左右的癫痫患者,其中500~600万是活动性癫痫患者,同时每年新增加癫痫患者约40万。
拉考沙胺具有双重抗癫痫作用机制:一方面,可选择性地增强电压促进钠通道缓慢失活,使过度兴奋神经元细胞膜稳定,以发挥镇静作用,并在不影响生理功能的情况下减少通道的长期可用性;另一方面,调节反应介导蛋白-2(crmp-2),而调节反应介导蛋白-2(crmp-2)可减慢、甚至阻止癫痫发作以及减轻糖尿病的神经性疼痛,达到抗惊厥的效果。
拉考沙胺有可能成为首个可干预癫痫发生和进展的药物。多个研究表明对耐药性癫痫、现有药物未能控制的癫痫部分发作患者疗效显著,同时研究证实拉考沙胺作为单药抗癫痫治疗部分性癫痫效果不劣于现有一线抗癫痫用药。拉考沙胺具有良好的药代动力学特性,如快速吸收、不受食物影响的高口服生物利用度(100%)、低个体间和个体内差异的线性剂量比例药代动力学。拉考沙胺有以下四点优势:非肝药酶诱导或者抑制作用,与多数抗癫痫药物(左乙拉西坦、卡马西平、拉莫三嗪、加巴喷丁、托吡酯、丙戊酸钠、苯妥英钠)具有较好的协同或相加作用;对现有药物无法控制症状的癫痫患者有较好的疗效;患者耐受性好,无认知功能损害弱,副作用小、更安全,尤其对于老年患者或存在基础疾病的患者。
现阶段,国内外报道的拉考沙胺的制备方法很多,按照起始物料划分主要有以下几种:
(1)专利us6048899中报道的制备方法以d-丝氨酸为起始原料,经过cbz氨基保护、氯甲酸乙酯成混酸酐后与苄胺缩合、碘甲烷在氧化银作用下甲基化、pd/c催化剂下脱保护和醋酸酐乙酰化等反应过程后合成拉考沙胺,是较为经典的合成方法,也是目前常用的产业化工艺路线,其反应路线大致如下所示:
注:cbz-为苄氧羰基,ibcf为氯甲酸异丙酯,nmm为n-甲基吗啉
该路线第二步使用了氯甲酸异丙酯做成混酸酐后再与苄胺进行缩合;甲基化使用了碘甲烷、氧化银,价格昂贵;脱氨基保护是使用了钯碳还原,生产成本较高,原子利用率低。
(2)专利us20080027137中报道的制备方法是以d-丝氨酸为起始物料,经过boc氨基保护、硫酸二甲酯甲基化、氯甲酸乙酯成混酸酐后与苄胺缩合、盐酸下脱保护和醋酸酐乙酰化等反应过程后合成拉考沙胺,是较为经典的合成方法,也是目前常用的产业化工艺路线,其反应路线大致如下所示:
注:boc-为碳酸叔丁酯
该路线第二步使用了硫酸二甲酯作为甲基化试剂,硫酸二甲酯为剧毒品,且反应完后需要用大量的酸去中和,产生大量废水;第四步脱保护使用了大量的盐酸,反应完后需要大量的碱去中和,产生大量废水,环保处理成本很高。
(3)以苄氧基甲基环氧乙烷为起始原料,经过开环醚化,叠氮化、氢化、boc保护、羟基氧化、酰胺化,最后再去保护,乙酰化合成拉科酰胺,该路线长而复杂,收率低,不适于产业化生产,其反应路线大致如下所示:
同样的,以2-乙烯基环氧乙烷为原料经过5步反应,或者以卤代甲基环氧丙烷为原料经过7步反应合成拉科酰胺,均存在路线过长,步骤复杂的问题,其反应路线大致如下所示:
(3)以n-苄基丙烯酰胺为原料,经过烯烃卤代、甲氧基化、氨化,最后乙酰化合成拉科酰胺,也是较为常用的产业化方法,但是该方法中用到液溴有毒有害物质,极易过敏,其反应路线大致如下所示:
技术实现要素:
本发明所要解决的技术问题是为了克服现有的拉考沙胺制备方法的生产成本高、原子利用率低、使用剧毒试剂、生成大量废水即环境污染大、路线复杂收率低等缺陷,而提供了一种拉考沙胺的合成方法,该方法的工艺路线操作简单,该反应路线具有原子的经济性高,避免保护基碳酸叔丁酯的使用,避免甲基化试剂硫酸二甲酯的使用,易制毒品氯甲酸乙酯的使用,收率高等优点,适于工业化生产。
一种拉考沙胺的合成方法,其包括如下步骤:a1.将式ⅱ化合物和原甲酸三甲酯加成反应生成式ⅲ化合物;a2.将式ⅲ化合物在催化剂和配体的作用下和h2发生不对称氢化反应生成式ⅳ化合物;a3.将式ⅳ化合物在酸的作用下和叠氮钠发生schmidt重排反应生成式ⅴ化合物;a4.将式ⅴ化合物在甲醇钠碱性的条件下和苄胺(bnnh2)发生胺化反应生成式ⅰ拉考沙胺;
其中,所述r是c1-c4烷基,优选甲基、乙基、丙基、异丙基、正丁基、异丁基中的一种。
在所述的拉考沙胺的合成方法中,所述步骤a1为将式ii化合物、醋酸酐和原甲酸三甲酯加入到反应瓶中,搅拌升温至回流,反应过程体系颜色逐渐变成深红色,反应完毕,减压蒸馏得式iii化合物。
在所述的拉考沙胺的合成方法中,所述步骤a2为在氢化罐中加入a1中式iii化合物、催化剂、配体和溶剂,经过反应之后处理得到式iv化合物,不经提纯,直接进入下一步骤。
在所述的拉考沙胺的合成方法中,所述步骤a3为在反应瓶中,加入a2中式iv化合物、酸和溶剂,在一定温度和搅拌的条件下,分批加入叠氮化钠,反应完毕后,得式v化合物。
在所述的拉考沙胺的合成方法中,所述步骤a4为在反应瓶中,加入a3中式v化合物、苄胺、27%甲醇钠溶液和溶剂,搅拌升温至回流,取样检测,反应完毕,经过处理后,得拉考沙胺(式i化合物)粗品湿品。
在所述的拉考沙胺的合成方法中,步骤a5为在反应瓶中,加入步骤a4的拉考沙胺(式i化合物)粗品湿品全量和溶剂,经过处理后,得拉考沙胺(式i化合物)干品。
在所述的拉考沙胺的合成方法中,所述步骤a1中式ii和原甲酸三甲酯的摩尔比为1:(1~2.5),较佳地为1:(1.5~2)。
在所述的拉考沙胺的合成方法中,所述步骤a2中,所述催化剂为二碘代(对-伞花烃)钌(ii)二聚物。
在所述的拉考沙胺的合成方法中,所述步骤a2中,所述配体为(αs,αs)-2,2’-双α-n,n-二甲基氨基苯基甲基)-(r,r)-1,1’-双[二(3,5-二甲基-4-甲氧基苯基)膦]二茂铁。
在所述的拉考沙胺的合成方法中,所述步骤a2中,所述溶剂为酯类溶剂和醇类溶剂,所述酯类溶剂选自乙酸乙酯、乙酸甲酯、乙酸异丙酯中的一种或两种,所述醇类溶剂选自乙醇、甲醇、异丙醇中的一种或两种。
在所述的拉考沙胺的合成方法中,所述步骤a2中,反应温度为10~50℃,优选20~30℃,反应时间为5~30小时,优选10~20小时。
在所述的拉考沙胺的合成方法中,所述步骤a2中每当量的式iii化合物参与反应,需要的催化剂为0.0001~0.005当量,较佳地催化剂的摩尔量为0.001~0.002当量。所述步骤a2中,催化剂和配体的摩尔比为1:(2~5)。氢气的压力为1~5mpa,较佳地为2~3mpa。
在所述的拉考沙胺的合成方法中,所述步骤a3中,所述酸选自硫酸、多聚磷酸、对甲苯磺酸、甲磺酸、醋酸中的一种或几种。
在所述的拉考沙胺的合成方法中,所述步骤a3中,所述溶剂为甲苯、二甲苯、二氯甲烷、三氯甲烷中的一种或几种。
在所述的拉考沙胺的合成方法中,所述步骤a3中,反应温度为10~50℃,优选20~30℃,反应时间为1~8小时,优选1~4小时。
在所述的拉考沙胺的合成方法中,所述步骤a3中每当量的式iv化合物参与反应,酸的加入量为1~3当量,叠氮化钠的加入量为1~3当量。
在所述的拉考沙胺的合成方法中,所述步骤a4中,所述溶剂为低级醇类溶剂,优选为无水乙醇、无水甲醇、无水异丙醇中的一种。
在所述的拉考沙胺的合成方法中,所述步骤a4中每当量的式v化合物参与反应,苄胺的加入量为1~1.5当量,较佳地为1~1.2当量。
在所述的拉考沙胺的合成方法中,所述步骤a5中,所述溶剂为酯类溶剂,优选为乙酸甲酯、乙酸乙酯、乙酸丙酯中的一种。
本领域技术人员知晓,在不违背本领域常识的基础上,上述各条件均可任意组合即得本发明各较佳实施例。
有益效果:与现有技术相比,本发明经过加成,氢化、重排、胺化合成拉考沙胺,本发明提供的合成方法的工艺路线操作简单,该反应路线具有原子的经济性高,避免保护基碳酸叔丁酯的使用,避免甲基化试剂硫酸二甲酯的使用,易制毒品氯甲酸乙酯的使用,收率高等优点,适于工业化生产。
附图说明
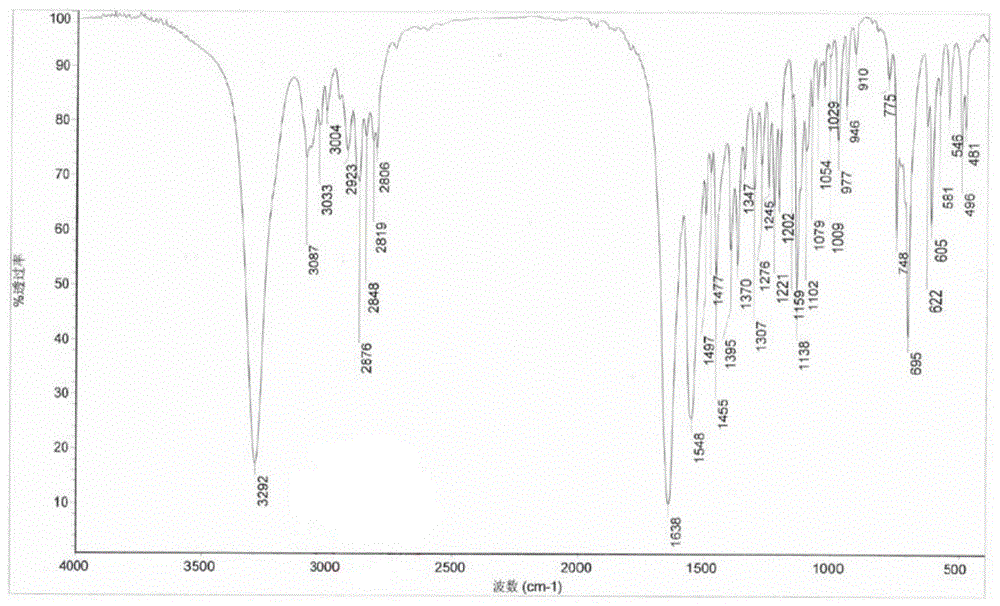
图1拉考沙胺的红外ir谱图;
图2拉考沙胺的nmr-13c谱图;
图3拉考沙胺的nmr-1h谱图。
具体实施方式
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围内。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
实施例1
1.(e)-2-(甲氧基亚甲基)-3-氧代丁酸乙酯,式iii化合物的合成
在250ml反应瓶中,加入50g乙酰乙酸乙酯,94g醋酸酐和65g原甲酸三甲酯,搅拌升温至回流,反应过程体系颜色逐渐变成深红色。反应完毕,减压蒸馏得61.5g油状缩合物,收率92.97%,1hnmr(500mhz,cdcl3),8.22(s,1h,ch),4.20(m,2h,ch2),3.81(s,3h,och3),2.27(s,3h,coch3),1.29(t,3h,ch3)。
2.(r)-2-(甲氧基甲基)-3-氧代丁酸乙酯,式iv化合物的合成
在氢化罐中加入30g式iii化合物,0.3g催化剂(二碘代(对-伞花烃)钌(ii)二聚物)、0.1g配体((αs,αs)-2,2’-双α-n,n-二甲基氨基苯基甲基)-(r,r)-1,1’-双[二(3,5-二甲基-4-甲氧基苯基)膦]二茂铁(=mandyphossl-m004-2)和150g异丙醇,用氮气置换氢化罐中的空气,重复此操作三次后,用氢气置换氢化罐中的氮气,重复此操作2次后充入2mpa氢气,20℃下搅拌10h。然后用氮气置换氢化罐中的氢气,重复此操作三次后,减压浓缩异丙醇后,加入水和二氯甲烷,萃取分液,二氯甲烷层干燥,减压浓缩,得到28.3g油状物式iv化合物,收率约94.02%,纯度99.05%,式iv化合物:异构体=96.8:3.2,不经提纯,直接进入下一工序。
1hnmr(500mhz,cdcl3),4.10-4.25(m,2h,ch2),3.78-3.91(m,1h,ch),3.52(m,1h,ch),3.29(s,3h,och3),2.28(s,3h,coch3),1.29(t,3h,ch3)
3.n-乙酰-o-甲基-l-丝氨酸乙酯,式v化合物的合成
在500ml反应瓶中,加入50g式iv化合物,31g多聚磷酸和300g三氯甲烷,控温20℃,搅拌下分批加入21g叠氮化钠,加毕,室温搅拌反应4h,取样检测。反应完毕,加水萃取分液,三氯甲烷层用用硫酸钠干燥后,浓缩得49.64g式v化合物,收率91.41%,纯度94.9%。
1hnmr(500mhz,cdcl3),4.54(t,1h,ch),4.39(t,2h,ch2),3.59-3.68(dd,2h,ch2),3.34(s,3h,och3),2.00(s,3h,coch3),1.29(t,3h,ch3)
4.(r)-2-乙酰氨基-n-苄基-3-甲氧基丙酰胺,拉考沙胺(式i化合物)粗品湿品的合成
在500ml反应瓶中,加入50g式v化合物,30g苄胺、10g27%甲醇钠溶液和200g无水甲醇,搅拌升温至回流3h,取样检测,反应完毕,减压浓缩后,加入200g乙酸甲酯升温溶解后,降温析晶,过滤得拉考沙胺(式i化合物)粗品湿品。
5.(r)-2-乙酰氨基-n-苄基-3-甲氧基丙酰胺,拉考沙胺(式i化合物)干品的合成
在500ml反应瓶中,加入上述粗品湿品全量和200g乙酸甲酯,升温回流溶解后,加入活性炭脱色,过滤,滤液降温析晶,过滤得到拉考沙胺精品湿品,控温50℃,减压干燥7h,收料得56.82g干品,总收料86.09%,纯度99.96%,异构体杂质a<0.01%。
实施例2
1.(e)-2-(甲氧基亚甲基)-3-氧代丁酸乙酯,式(iii)的合成
在250ml反应瓶中,加入50g乙酰乙酸乙酯,94g醋酸酐和65g原甲酸三甲酯,搅拌升温至回流,反应过程体系颜色逐渐变成深红色。反应完毕,减压蒸馏得61.5g油状缩合物,收率92.97%,1hnmr(500mhz,cdcl3),8.22(s,1h,ch),4.20(m,2h,ch2),3.81(s,3h,och3),2.27(s,3h,coch3),1.29(t,3h,ch3)。
2.(r)-2-(甲氧基甲基)-3-氧代丁酸乙酯,式(iv)的合成
在氢化罐中加入30g化合物iii,0.3g催化剂(二碘代(对-伞花烃)钌(ii)二聚物)、0.1g配体((αs,αs)-2,2’-双α-n,n-二甲基氨基苯基甲基)-(r,r)-1,1’-双[二(3,5-二甲基-4-甲氧基苯基)膦]二茂铁(=mandyphossl-m004-2)和150g乙醇,用氮气置换氢化罐中的空气,重复此操作三次后,用氢气置换氢化罐中的氮气,重复此操作2次后充入2mpa氢气,30℃下搅拌20h。然后用氮气置换氢化罐中的氢气,重复此操作三次后,减压浓缩甲醇后,加入水和二氯甲烷,萃取分液,二氯甲烷层干燥,减压浓缩,得到29.37g油状物化合物iv,收率约97.57%,纯度99.05%,化合物iv:异构体=96.8:3.2,不经提纯,直接进入下一工序。
1hnmr(500mhz,cdcl3),4.10-4.25(m,2h,ch2),3.78-3.91(m,1h,ch),3.52(m,1h,ch),3.29(s,3h,och3),2.28(s,3h,coch3),1.29(t,3h,ch3)
3.n-乙酰-o-甲基-l-丝氨酸乙酯,式(v)的合成
在500ml反应瓶中,加入50g化合物iv,31g甲磺酸和300g二甲苯,控温30℃,搅拌下分批加入21g叠氮化钠,加毕,室温搅拌反应4h,取样检测。反应完毕,加水萃取分液,二甲苯层用硫酸钠干燥后,浓缩得45.16g化合物v,收率83.15%,纯度94.9%。
1hnmr(500mhz,cdcl3),4.54(t,1h,ch),4.39(t,2h,ch2),3.59-3.68(dd,2h,ch2),3.34(s,3h,och3),2.00(s,3h,coch3),1.29(t,3h,ch3)
4.(r)-2-乙酰氨基-n-苄基-3-甲氧基丙酰胺,拉考沙胺(式i化合物)粗品湿品的合成
在500ml反应瓶中,加入50g化合物v,30g苄胺、10g27%甲醇钠溶液和200g无水甲醇,搅拌升温至回流3h,取样检测,反应完毕,减压浓缩后,加入200g乙酸丙酯升温溶解后,降温析晶,过滤得拉考沙胺(式i化合物)粗品湿品。
5.(r)-2-乙酰氨基-n-苄基-3-甲氧基丙酰胺,拉考沙胺(式i化合物)干品的合成
在500ml反应瓶中,加入上述粗品湿品全量和200g乙酸丙酯,升温回流溶解后,加入活性炭脱色,过滤,滤液降温析晶,过滤得到拉考沙胺精品湿品,控温60℃,减压干燥5h,收料得56.89g干品,总收料86.20%,纯度99.96%,异构体杂质a<0.01%。
实施例3
1.(e)-2-(甲氧基亚甲基)-3-氧代丁酸乙酯,式(iii)的合成
在250ml反应瓶中,加入50g乙酰乙酸乙酯,94g醋酸酐和65g原甲酸三甲酯,搅拌升温至回流,反应过程体系颜色逐渐变成深红色。反应完毕,减压蒸馏得61.5g油状缩合物,收率92.97%,1hnmr(500mhz,cdcl3),8.22(s,1h,ch),4.20(m,2h,ch2),3.81(s,3h,och3),2.27(s,3h,coch3),1.29(t,3h,ch3)。
2.(r)-2-(甲氧基甲基)-3-氧代丁酸乙酯,式(iv)的合成
在氢化罐中加入30g化合物iii,0.3g催化剂(二碘代(对-伞花烃)钌(ii)二聚物)、0.1g配体((αs,αs)-2,2’-双α-n,n-二甲基氨基苯基甲基)-(r,r)-1,1’-双[二(3,5-二甲基-4-甲氧基苯基)膦]二茂铁(=mandyphossl-m004-2)和150g无水甲醇,用氮气置换氢化罐中的空气,重复此操作三次后,用氢气置换氢化罐中的氮气,重复此操作2次后充入2mpa氢气,25℃下搅拌15h。然后用氮气置换氢化罐中的氢气,重复此操作三次后,减压浓缩甲醇后,加入水和二氯甲烷,萃取分液,二氯甲烷层干燥,减压浓缩,得到30.1g油状物化合物iv,收率约100%,纯度99.05%,化合物iv:异构体=96.8:3.2,不经提纯,直接进入下一工序。
1hnmr(500mhz,cdcl3),4.10-4.25(m,2h,ch2),3.78-3.91(m,1h,ch),3.52(m,1h,ch),3.29(s,3h,och3),2.28(s,3h,coch3),1.29(t,3h,ch3)
3.n-乙酰-o-甲基-l-丝氨酸乙酯,式(v)的合成
在500ml反应瓶中,加入50g化合物iv,31g浓硫酸和300g二氯甲烷,控温25℃,搅拌下分批加入21g叠氮化钠,加毕,室温搅拌反应2h,取样检测。反应完毕,加水萃取分液,二氯甲烷层用用硫酸钠干燥后,浓缩得50.5g化合物v,收率92.98%,纯度94.9%。
1hnmr(500mhz,cdcl3),4.54(t,1h,ch),4.39(t,2h,ch2),3.59-3.68(dd,2h,ch2),3.34(s,3h,och3),2.00(s,3h,coch3),1.29(t,3h,ch3)
4.(r)-2-乙酰氨基-n-苄基-3-甲氧基丙酰胺,拉考沙胺(式i化合物)粗品湿品的合成
在500ml反应瓶中,加入50g化合物v,30g苄胺、10g27%甲醇钠溶液和200g无水乙醇,搅拌升温至回流3h,取样检测,反应完毕,减压浓缩后,加入200g乙酸乙酯升温溶解后,降温析晶,过滤得拉考沙胺粗品湿品。
5.(r)-2-乙酰氨基-n-苄基-3-甲氧基丙酰胺,拉考沙胺(式i化合物)干品的合成
在500ml反应瓶中,加入上述粗品湿品全量和200g乙酸乙酯,升温回流溶解后,加入活性炭脱色,过滤,滤液降温析晶,过滤得到拉考沙胺精品湿品,控温55℃,减压干燥6h,收料得60g干品,总收料90.91%,纯度99.96%,异构体杂质a<0.01%。
以上所述,仅是本申请的几个实施例,并非对本申请做任何形式的限制,任何本专业的技术人员在不脱离本申请技术方案的范围内,利用上述揭示的技术内容做出些许的变动或修饰均等效本专利,属于本技术方案保护的范围内。
- 还没有人留言评论。精彩留言会获得点赞!