N-乙烯基吡咯烷酮的合成方法与流程
本发明涉及精细化学品合成领域,具体而言,涉及一种n-乙烯基吡咯烷酮的合成方法。
背景技术:
n-乙烯基吡咯烷酮(简称nvp)主要用于生产聚乙烯吡咯烷酮(简称pvp),pvp是一种非离子型水溶性高分子精细化学品,它不仅具有优异的溶解性、化学稳定性、成膜性、生理惰性、黏结能力和保护胶作用,而且还可以与许多无机、有机、高分子化合物结合形成多种具有独特功能的、其他化合物不可比拟的新型精细化学品。由于这些特性,pvp在医药卫生、日用化工、颜料涂料、办公文具、饮料与食品等行业有广泛的应用。
单体nvp的合成是pvp产业链的关键部分,乙炔法(又称reppe法)是工业生产nvp的主要工艺。乙炔法指通过乙炔和2-吡咯烷酮在一定温度、压力下,并以吡咯烷酮钾等强碱为催化剂发生亲和加成反应从而生成nvp。nvp的合成,从早期的学术研究到现在成熟的工业生产,一般都采用高压釜进行。一方面,采用高压釜生产有一些问题仍需解决:首先该反应是气液两相反应,反应速率受气液传质控制,反应过程中很大程度上依赖于搅拌转速,反应速率低直接导致反应时间很长(3~5h);其次该过程属于间歇操作,操作可控性和产品均一性相对较差。另一方面,由于乙炔的易燃易爆性,乙炔的安全是工业生产时亟需解决的关键问题:其一,乙炔氧化爆炸的范围比其他气体都宽,和空气混合物的爆炸极限范围在2.2~82%(v),并且所需点火能量小,含7.7%乙炔的混合气最低引爆能量为1.9×10-5j(仅为未熄灭的烟火能量级);其二,乙炔极易发生分解爆炸,引起乙炔分解爆炸的典型因素有受热、加压、静电、铜、银、铁锈等。
有鉴于此,特提出本发明。
技术实现要素:
本发明的目的在于提供一种n-乙烯基吡咯烷酮的合成方法,该方法采用液相循环方式,使液体原料2-吡咯烷酮达到高转化率,采用新型反应器微通道反应器使产物nvp达到高选择性,并可确保乙炔使用的本质安全,强化传质传热,实现连续化生产,严格保证操作可控性和产品一致性。
为了实现本发明的上述目的,特采用以下技术方案:
本发明提供了一种n-乙烯基吡咯烷酮的合成方法,包括:将乙炔与含催化剂的2-吡咯烷酮液体通入微通道反应器中进行反应,得到n-乙烯基吡咯烷酮;
其中:含催化剂的2-吡咯烷酮液体中催化剂的含量为2.2-10.5wt%;乙炔与含催化剂的2-吡咯烷酮液体的体积流量比为(32-270):1;微通道反应器中单通道的内径小于3mm。
作为进一步优选的技术方案,微通道反应器中单通道的内径为0.5-1.5mm。
作为进一步优选的技术方案,催化剂的含量为5.4-7.4wt%;
优选地,所述体积流量比为(50-200):1。
作为进一步优选的技术方案,反应温度为140-180℃;
优选地,反应压力为0.6-1.2mpa。
作为进一步优选的技术方案,乙炔以含有乙炔的混合气的形式提供,乙炔占混合气的体积比为40-90%,优选为50-70%;
优选地,所述混合气中还包括氮气和/或惰性气体。
作为进一步优选的技术方案,所述催化剂包括2-吡咯烷酮的碱金属盐;
优选地,所述催化剂包括吡咯烷酮锂、吡咯烷酮钠、吡咯烷酮钾或吡咯烷酮铯中的至少一种,优选为吡咯烷酮钾。
作为进一步优选的技术方案,含催化剂的2-吡咯烷酮液体的制备方法包括:将碱金属氢氧化物和2-吡咯烷酮液体混合后经减压蒸馏,得到含催化剂的2-吡咯烷酮液体;
优选地,碱金属氢氧化物和2-吡咯烷酮液体的质量比为(1-5):100,优选为(2.5-3.5):100。
作为进一步优选的技术方案,在反应前还包括将乙炔和含催化剂的2-吡咯烷酮液体进行混合的步骤;
优选地,在反应前还包括将乙炔和含催化剂的2-吡咯烷酮液体进行预热的步骤;
优选地,在反应前还包括将乙炔和含催化剂的2-吡咯烷酮液体依次进行混合和预热的步骤;
优选地,预热温度为120-135℃。
作为进一步优选的技术方案,在反应后还包括对产物依次经任选的冷却、气液分离和气液分离所得液相混合物被重新送入微通道反应器中进行再次反应,直至液相混合物中nvp收率达到要求的步骤,最后对液相混合物进行分离和提纯,得到n-乙烯基吡咯烷酮。
作为进一步优选的技术方案,所述方法包括:
(a)将质量比为(1-5):100的碱金属氢氧化物和2-吡咯烷酮液体混合后经减压蒸馏,得到含催化剂的2-吡咯烷酮液体;
(b)将含乙炔的混合气和含催化剂的2-吡咯烷酮液体依次进行混合和预热;乙炔占混合气的体积比为40-90%,预热温度为120-135℃;
(c)将含乙炔的混合气和含催化剂的2-吡咯烷酮液体通入微通道反应器中进行反应;反应温度为140-180℃,反应压力为0.6-1.2mpa;
(d)反应后对产物依次经冷却、气液分离和气液分离所得液相混合物被重新送入微通道反应器中进行再次反应,直至液相混合物中nvp收率达到要求,最后对液相混合物进行分离和提纯,得到n-乙烯基吡咯烷酮。
与现有技术相比,本发明的有益效果为:
本发明提供的合成方法中采用了特定单通道内径的微通道反应器进行反应原料之间的反应,该内径非常小,具有阻火器效应,可以大大降低乙炔发生分解爆炸的风险,而且微通道反应器还具有较高的比表面积和较短的物料扩散距离,其中的质量和热量可实现快速传递和精确控制,从而强化气液传质传热、加快原料之间的反应速率、实现放热反应的等温操作,出现热点或超温的概率大大降低,操作可控性和产品一致性较高。此外,相比于高压釜的间歇操作,微通道反应器可实现连续化生产。
同时,该方法中还采用了特定的催化剂含量和特定的体积流量比,从而使2-吡咯烷酮液相反应物相对于乙炔气体来说是大大过量的,液相为连续相,而气体成为分散的气柱,气液流型为典型的taylorflow,并在催化剂的作用下,使乙炔快速完全转化,进一步保证乙炔的使用安全性。
附图说明
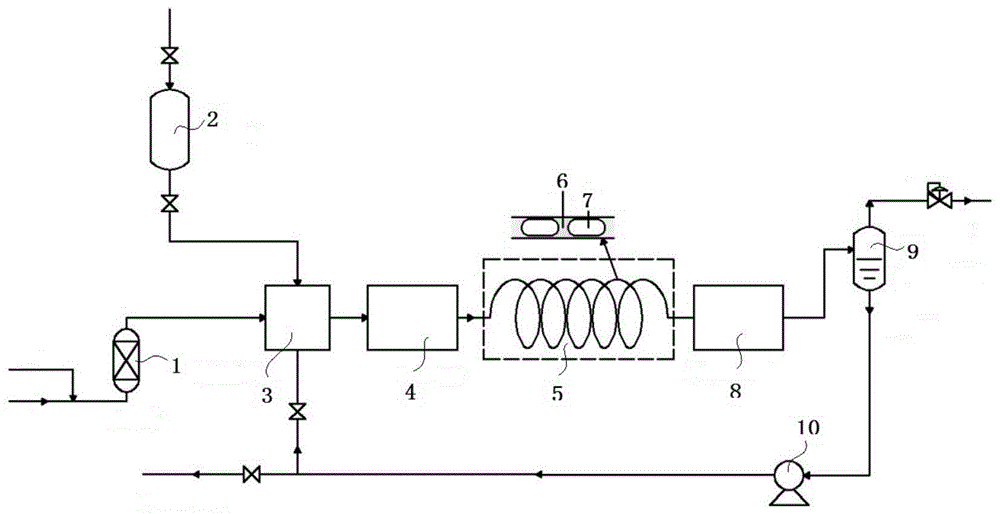
图1为实施例1中n-乙烯基吡咯烷酮的合成方法的工艺流程示意图。
图标:1-干燥柱;2-缓冲罐;3-微混合器;4-快速加热器;5-微通道反应器;6-液柱;7-气柱;8-快速冷却器;9-气液分离器;10-循环泵。
具体实施方式
下面将结合实施例对本发明的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限制本发明的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。
根据本发明的一个方面,提供了一种n-乙烯基吡咯烷酮的合成方法,包括:将乙炔与含催化剂的2-吡咯烷酮液体通入微通道反应器中进行反应,得到n-乙烯基吡咯烷酮;
其中:含催化剂的2-吡咯烷酮液体中催化剂的含量为2.2-10.5wt%;乙炔与含催化剂的2-吡咯烷酮液体的体积流量比为(32-270):1;微通道反应器中单通道的内径小于3mm。
上述合成方法中采用了特定单通道内径的微通道反应器进行反应原料之间的反应,该内径非常小,具有阻火器效应,可以大大降低乙炔发生分解爆炸的风险,而且微通道反应器还具有较高的比表面积和较短的物料扩散距离,其中的质量和热量可实现快速传递和精确控制,从而强化气液传质传热、加快原料之间的反应速率、实现放热反应的等温操作,出现热点或超温的概率大大降低,操作可控性和产品一致性较高。此外,相比于高压釜的间歇操作,微通道反应器可实现连续化生产。
同时,该方法中还采用了特定的催化剂含量和特定的体积流量比,从而使2-吡咯烷酮液相反应物相对于乙炔气体来说是大大过量的,液相为连续相,而气体成为分散的气柱,气液流型为典型的taylorflow,并在催化剂的作用下,使乙炔快速完全转化,进一步保证乙炔的使用安全性。
上述“催化剂的含量”是指催化剂占含催化剂的2-吡咯烷酮液体的质量百分比。
上述催化剂的含量典型但非限制性的为2.2wt%、2.5wt%、3wt%、3.5wt%、4wt%、4.5wt%、5wt%、5.5wt%、6wt%、6.5wt%、7wt%、7.5wt%、8wt%、8.5wt%、9wt%、9.5wt%、10wt%或10.5wt%。上述体积流量比典型但非限制性的为32:1、40:1、50:1、60:1、70:1、80:1、90:1、100:1、120:1、140:1、160:1、180:1、200:1、220:1、240:1、250:1或270:1。上述体积流量比较为合理,能够使乙炔相对于2-吡咯烷酮大大过量,有效保证乙炔的使用安全性。体积流量比过小则反应速率较慢,而体积流量比过大则乙炔的使用安全性较差。
上述单通道的内径例如可以为0.1、0.2、0.3、0.4、0.5、0.6、0.7、0.8、0.9、1、1.2、1.3、1.4、1.5、1.7、2、2.2、2.4、2.6、2.8或2.9mm。
上述微通道反应器的材质可选为316l不锈钢或哈氏合金等,形式可以为列管式反应器。
在一种优选的实施方式中,微通道反应器中单通道的内径为0.5-1.5mm。经大量试验发现,当单通道的内径在以上范围内时,气液传质的效果更好,能够在有效消除乙炔安全隐患的前提下,加快反应速率和乙炔的转化率,进而提高nvp的产率。
优选地,催化剂的含量为5.4-7.4wt%。当催化剂的含量在上述范围内时,有利于进一步加快乙炔和2-吡咯烷酮的反应速率。
优选地,所述体积流量比为(50-200):1。通过进一步优选体积流量比,能够在保证乙炔安全使用的情况下,增加一次反应所生成的nvp的量,有利于加快生产效率。
优选地,反应温度为140-180℃。上述反应温度典型但非限制性的为140、145、150、155、160、165、170、175或180℃。
优选地,反应压力为0.6-1.2mpa。上述反应压力典型但非限制性的为0.6、0.7、0.8、0.85、0.9、0.95、1、1.1或1.2mpa。
当反应温度和反应压力在上述范围内时,乙炔和2-吡咯烷酮的反应速率更快,乙炔的转化率更高。
在一种优选的实施方式中,乙炔以含有乙炔的混合气的形式提供,乙炔占混合气的体积比为40-90%,优选为50-70%。通过以含有乙炔的混合气的形式提供乙炔,能使乙炔在气体中的浓度得到进一步稀释,进一步保证乙炔使用的安全性。
优选地,所述混合气中还包括氮气和/或惰性气体。惰性气体包括氦(he)、氖(ne)、氩(ar)、氪(kr)、氙(xe)、氡(rn,放射性)、气奥(og,放射性,人造元素)。
在一种优选的实施方式中,所述催化剂包括2-吡咯烷酮的碱金属盐。
优选地,所述催化剂包括吡咯烷酮锂、吡咯烷酮钠、吡咯烷酮钾或吡咯烷酮铯中的至少一种。
优选地,含催化剂的2-吡咯烷酮液体的制备方法包括:将碱金属氢氧化物和2-吡咯烷酮液体混合后经减压蒸馏,得到含催化剂的2-吡咯烷酮液体。通过本方法可一次性得到含有催化剂的2-吡咯烷酮液体,工艺步骤少,简单易于操作。
优选地,碱金属氢氧化物和2-吡咯烷酮液体的质量比为(1-5):100,优选为(2.5-3.5):100。碱金属氢氧化物和2-吡咯烷酮的质量比典型但非限制性的为1:100、1.5:100、2:100、2.5:100、3:100、3.5:100、4:100、4.5:100或5:100。
在一种优选的实施方式中,在反应前还包括将乙炔和含催化剂的2-吡咯烷酮液体进行混合的步骤。通过该步骤,可在乙炔和2-吡咯烷酮反应之前将乙炔提前溶解于2-吡咯烷酮液体中,能够增强后续反应的平稳性。
优选地,在反应前还包括将乙炔和含催化剂的2-吡咯烷酮液体进行预热的步骤。通过将反应物提前进行预热,可减少其进入微通道反应器中的加热时间,加快反应进程,提高nvp收率。
优选地,在反应前还包括将乙炔和含催化剂的2-吡咯烷酮液体依次进行混合和预热的步骤。在反应前通过混合和预热,使反应物提前进入一定的反应前状态,有利于加快反应进程,提高在微通道反应器内前后段反应的一致性和平稳性。
优选地,预热温度为120-135℃。上述预热温度典型但非限制性的为120、125、130或135℃。
可选地,上述混合可在微混合器中进行,上述预热可在加热器中进行。
在一种优选的实施方式中,在反应后还包括对产物依次经任选的冷却、气液分离和气液分离所得液相混合物被重新送入微通道反应器中进行再次反应,直至液相混合物中nvp收率达到要求的步骤,最后对液相混合物进行分离和提纯,得到n-乙烯基吡咯烷酮。
本优选实施方式提供了一种连续化生产的工艺流程,包括:反应后对反应物进行任选的冷却和气液分离,气液分离所得气体为未反应的乙炔或未反应的混合气,可去往废气处理系统,气液分离后所得液相混合物为包括产物nvp和未反应完的2-吡咯烷酮的液相混合物,该混合物被重新送入(例如采用循环泵泵送)微通道反应器中进行再次反应,实现液相原料2-吡咯烷酮的循环利用,直至液相混合物中的nvp含量达到指定收率后,不再循环,最后对液相混合物进行分离和提纯,得到n-乙烯基吡咯烷酮。
应当理解的是,本实施方式中“任选的”仅指冷却的步骤是任选的,而其余步骤是必须要有的。因此,本优选实施方式包括以下两种情况:一、在反应后对产物依次经气液分离和气液分离所得液相混合物被重新送入微通道反应器中进行再次反应,直至液相混合物中nvp收率达到要求的步骤,最后对液相混合物进行分离和提纯,得到n-乙烯基吡咯烷酮;二、在反应后对产物依次经冷却、气液分离和气液分离所得液相混合物被重新送入微通道反应器中进行再次反应,直至液相混合物中nvp收率达到要求的步骤,最后对液相混合物进行分离和提纯,得到n-乙烯基吡咯烷酮。
上述“液相混合物中nvp收率”是指nvp的实际产量与nvp的理论产量(以液体原料2-吡咯烷酮计算)的比值。
在一种优选的实施方式中,所述方法包括:
(a)将质量比为(1-5):100的碱金属氢氧化物和2-吡咯烷酮液体混合后经减压蒸馏,得到含催化剂的2-吡咯烷酮液体;
(b)将含乙炔的混合气和含催化剂的2-吡咯烷酮液体依次进行混合和预热;乙炔占混合气的体积比为40-90%,预热温度为120-135℃;
(c)将含乙炔的混合气和含催化剂的2-吡咯烷酮液体通入微通道反应器中进行反应;反应温度为140-180℃,反应压力为0.6-1.2mpa;
(d)反应后对产物依次经冷却、气液分离和气液分离所得液相混合物被重新送入微通道反应器中进行再次反应,直至液相混合物中nvp收率达到要求,最后对液相混合物进行分离和提纯,得到n-乙烯基吡咯烷酮。
本优选实施方式提供的方法工艺清晰、完整、科学,能够将乙炔尽可能完全转化,有效提高乙炔使用安全性,同时能实现nvp的连续化生产,提高2-吡咯烷酮的转化率,得到收率符合要求的nvp产品。
下面结合实施例和对比例对本发明做进一步详细的说明。
实施例1
一种n-乙烯基吡咯烷酮的合成方法,包括:
将0.6kg氢氧化钾和20kg2-吡咯烷酮置于反应装置中进行减压蒸馏,保持压力20mbar,温度120℃,反应2h后结束,此时该含催化剂的2-吡咯烷酮液体中水含量低于500ppm;
采用图1所示的工艺流程合成nvp,气体总流量为1.8nm3/h,其中乙炔和氮气的流量比为1:1,氮气和乙炔首先经过干燥柱1进行干燥,上述含催化剂的2-吡咯烷酮液体流量为6l/h,含催化剂的2-吡咯烷酮液体储存在缓冲罐2中,气体、液体以设定流量进入微混合器3,实现常温下的混合,之后进入快速加热器4,预热至120~135℃,然后进入恒定温度为160℃、压力为1mpa的微通道反应器5,微通道反应器5的单个通道中包含液柱6和气柱7,反应结束后的反应混合物经快速冷却器8和气液分离器9后可得到nvp粗产品。该微通道反应器单个通道内径为0.6mm,管长60m,共有100个相同的通道按照特定的方式排列。nvp粗产品经循环泵10后再次进入微混合器3中作为原料与含催化剂的2-吡咯烷酮液体和混合气混合然后反应,直至产品中nvp收率达标后,去往分离装置分离得到nvp;而经气液分离器9分离得到的尾气去往废气处理系统进行处理。
该操作条件下2-吡咯烷酮的单程转化率为14.1%,5次循环后2-吡咯烷酮的总转化率为65.0%,5次循环的总反应时间为10min,nvp收率为65%,产物nvp的选择性为100.0%。
实施例2
一种n-乙烯基吡咯烷酮的合成方法,其中催化剂制备和工艺流程与实施例1相同。反应温度180℃,气体总流量为1.2nm3/h,上述含催化剂的2-吡咯烷酮液体流量为12l/h,微通道反应器单个通道内径更换为0.5mm,管长40m的316l不锈钢毛细管,其他反应条件不变。
该操作条件下2-吡咯烷酮的单程转化率为7.5%,8次循环后2-吡咯烷酮的总转化率为57.3%,8次循环的总反应时间为8min,nvp收率为57%,产物nvp的选择性为99.5%。
实施例3
一种n-乙烯基吡咯烷酮的合成方法,包括:将乙炔与含吡咯烷酮钠的2-吡咯烷酮液体通入微通道反应器中进行反应,得到n-乙烯基吡咯烷酮;
其中:含吡咯烷酮钠的2-吡咯烷酮液体中吡咯烷酮钠的含量为2.2wt%;乙炔与含吡咯烷酮钠的2-吡咯烷酮液体的体积流量比为32:1;微通道反应器中单通道的内径为0.9mm;反应温度为200℃,反应压力为0.7mpa。
实施例4-6
一种n-乙烯基吡咯烷酮的合成方法,与实施例3不同的是,实施例4-6中,所述体积流量比分别为270:1、50:1和200:1。
实施例5-6中的体积流量比在本发明优选范围内。
实施例7-9
一种n-乙烯基吡咯烷酮的合成方法,与实施例6不同的是,实施例7-9中,单通道的内径分别为0.3、0.5和0.6mm。
实施例7-9中单通道的内径在本发明优选范围内。
实施例10-12
一种n-乙烯基吡咯烷酮的合成方法,与实施例9不同的是,实施例10-12中,反应温度分别为140、160和180℃。
实施例10-12中反应温度在本发明优选范围内。
实施例13-15
一种n-乙烯基吡咯烷酮的合成方法,与实施例12不同的是,实施例13-15中,反应压力分别为0.8、0.9或1mpa。
实施例13-15中反应压力在本发明优选范围内。
实施例16-18
一种n-乙烯基吡咯烷酮的合成方法,与实施例15不同的是,实施例16-18中,乙炔以含有乙炔的混合气的形式提供,乙炔占混合气的体积比分别为40%、60%和90%。
实施例19-21
一种n-乙烯基吡咯烷酮的合成方法,与实施例17不同的是,实施例19-21中吡咯烷酮钠的含量分别为5.4wt%、6wt%和7.4wt%。
实施例19-21中吡咯烷酮钠的含量在本发明优选范围内。
经试验验证,实施例3-21中的反应时间均在分钟级别,反应速率较快,且产物nvp的选择性均在99%以上。
对比例1
采用us4873336a实施例5的方法制备n-乙烯基吡咯烷酮,其中koh和2-吡咯烷酮质量比为1.2:100,反应温度160℃,乙炔分压0.68mpa(未给总压),反应8h,2-吡咯烷酮转化率为75%,n-乙烯基吡咯烷酮选择性为90%。
可见,对比例1的反应时间过长,n-乙烯基吡咯烷酮选择性较差。
需要说明的是:
符号说明:2-p指2-吡咯烷酮,nvp指n-乙烯基吡咯烷酮,mol指物质的量;收率=转化率×选择性。
尽管已用具体实施例来说明和描述了本发明,然而应意识到,在不背离本发明的精神和范围的情况下可以作出许多其它的更改和修改。因此,这意味着在所附权利要求中包括属于本发明范围内的所有这些变化和修改。
- 还没有人留言评论。精彩留言会获得点赞!