用于治疗黑素瘤的放射性化合物及其用途的制作方法
本申请要求于2018年6月28日提交的韩国专利申请no.2018-0074766的优先权,并且该申请的全部内容通过引用合并于此。
本发明涉及一种新型化合物及其用途,更具体地,涉及一种用于治疗黑素瘤的新型放射性化合物及其用途。
背景技术:
恶性黑色素瘤由于其高度的全身转移而被称为最致命的癌症之一。恶性黑色素瘤占所有皮肤癌的5%,但其占皮肤癌相关死亡的50%以上。此外,该疾病的发病率在过去的二十年中翻了一番,并且还在稳步增加。迄今为止,还尚未开发出用于黑色素瘤的有效治疗方法。然而,早期诊断和准确确定疾病的阶段被认为是提高恶性黑色素瘤患者存活率的重要途径。
最近,18f-n-[2-(二乙基氨基)乙基]-4-氟-苯甲酰胺(18f-fbza)被开发出来,并被报道了其作为靶向黑色素的pet造影剂用于检测转移性黑色素瘤的应用(ren等,j.nucl.med.50(10):1692-1699,2009)。然而在用于黑色素瘤检测的pet造影剂时,其原理为利用苯甲酰胺结构的化学转化来诱导黑色素瘤选择性摄取的原理,但是黑色素瘤的摄取率低并且图像质量差,因此存在需要改进的问题。此外,直到现在,黑素瘤的唯一治疗选择是手术切除病变并使用抗癌药物。然而,在手术时不仅存在很多位置限制,并且使用的药物具有复发风险的缺点,同时治疗反应的效率非常低。因此,通过恶性黑色素瘤的选择性摄取放射性化合物的放射疗法被认为是最有效的方法,但是尚未开发出靶向黑色素瘤的有效放射性药物。
技术实现要素:
技术问题
本发明旨在解决包括上述问题在内的各种问题,并且本发明的目的是提供具有改善的黑素瘤靶向能力的新型放射性治疗化合物。
本发明的另一个目的是提供一种用于治疗黑素瘤的药物组合物,其包含所述化合物。
然而,这些问题为示例性的,并且本发明的范围不限于此。
技术方案
在本发明的一个方面,提供了一种新型放射性化合物或其可接受的盐,其结构如式1或式2所示:
(在上式中,x1和x2各自独立地为碳或氮,其中至少一个为氮;l1不存在或为化学键,或为取代或未取代的具有1至20个碳原子的亚烷基、取代或未取代的具有6至14个碳原子的烯丙基、取代或未取代的具有1至20个碳原子的杂亚烷基,具有2至60个碳原子的(聚)亚烷基二醇,或一种或多种选自以下的接头
本发明的另一方面提供了一种用于治疗恶性黑色素瘤的药物组合物,其包含式1或2的化合物或其药学上可接受的盐作为活性成分。
有利效果
根据本发明的一个实施方案的新型放射性化合物及其药学上可接受的盐可用作治疗黑素瘤的治疗剂。根据本发明实施方案的放射性化合物不仅具有改善的针对黑素瘤的靶向能力,而且具有非常显著地抑制黑素瘤的生长的优点。但是,本发明的范围不限于上述效果。
附图说明
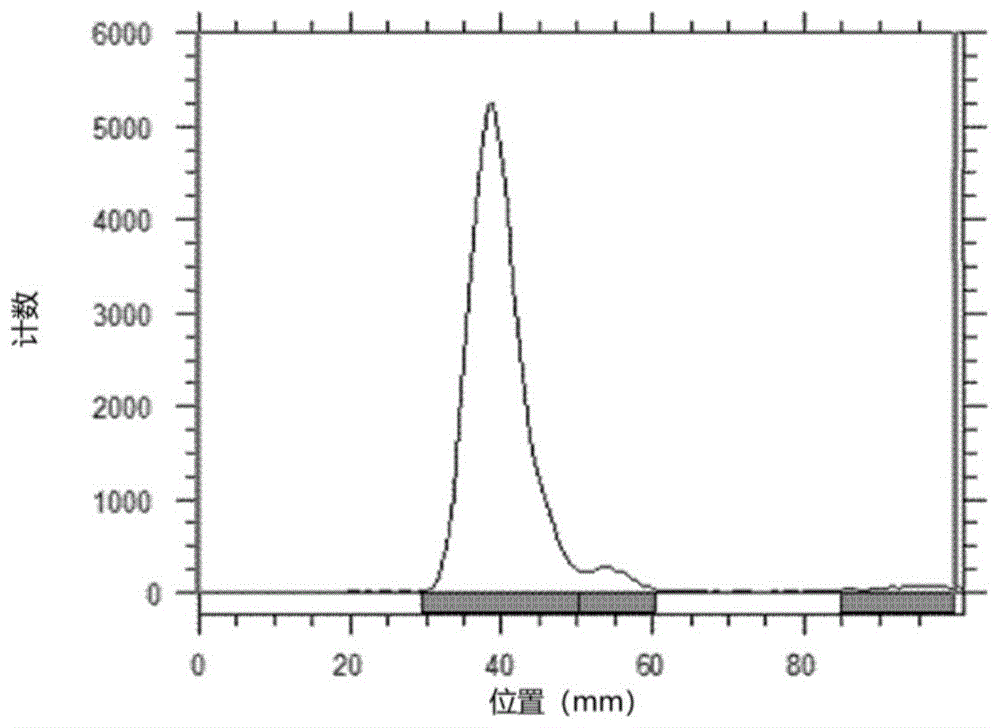
图1为示出了分析标记产率和放射化学纯度的结果的色谱图,该结果通过用放射性薄层色谱将根据本发明的实施方案的放射性化合物分离后得到。
图2为显示当将根据本发明的一个实施方案的177lu-dota-ncs-dmpy2施用于黑素瘤小动物模型时肿瘤生长随时间变化的图。
图3为显示当根据本发明的一个实施方案的177lu-dota-ncs-dmpy2施用于黑素瘤小动物模型时体重随时间的变化的图。
具体实施方式
本发明的一个方面提供了具有式1或2的结构的新型放射性化合物或其药学上可接受的盐。
(在上式中,x1和x2各自独立地为碳或氮,其中至少一个为氮;l1不存在或为化学键,或为取代或未取代的具有1至20个碳原子的亚烷基、取代或未取代的具有6至14个碳原子的烯丙基、取代或未取代的具有1至20个碳原子的杂亚烷基,具有2至60个碳原子的(聚)亚烷基二醇,或一种或多种选自以下的接头
在所述新型放射性化合物或其可接受的盐中,在式1和r1的情况下,和在式2的情况下,[m←l]部分是放射性同位素部分,并且l1是连接所述放射性同位素部分和黑色素瘤靶向部分的接头部分,并且不包括[m←l]部分和l1的r1或右结构与黑色素瘤靶部分对应。
在所述新型放射性化合物或其药学上可接受的盐中,接头l1可以是将螯合剂通过点击化学连接至黑素瘤靶向部分的模块化接头,所述点击化学例如叠氮化物-炔烃环加成反应,除了一般有机合成的、通过点击化学的炔烃-亚硝基反应,(炔烃-硝基环加成)反应,烯烃和叠氮化物[3+2]环加成反应,烯烃和四嗪逆需求diels-alder)反应,以及除了一般有机合成的、炔烃和四嗪光点击反应形成三唑或二苯并三唑并恶唑啉(dibenzotriazoloazocine)。模块化接头可包括一个或多个功能性接头,包括烃链连头(-[ch2]n-)或聚乙二醇(-[c2h4o]n-),其用作放射性同位素部分与黑素瘤靶向部分之间隔开一定距离的间隔基,及疏水部分(例如,
在所述新型放射性化合物或其药学上可接受的盐中,所述化学键可以是酯键、酰胺键、醚键、硫醚键、硫酯键或二硫键,并且l2可更优选为具有1至5个碳原子的亚烷基(-(ch2)n-,n为1至5的整数),最优选为亚乙基(-ch2ch2-)或亚丙基(-ch2ch2ch2-)。
本发明的另一方面提供了一种用于治疗黑素瘤的造影剂,其包含式1或2的化合物或其药学上可接受的盐作为活性成分。
式1的化合物可以通过以下方案制备:
另外,一旦合成了具有一般卤素元素而不是放射性卤素元素的放射性化合物,就可以使用含有放射性卤素元素的盐,例如80mbr和123i通过取代反应制备r1。备选地,也可以通过与具有放射性同位素卤素或放射性磷作为官能团(例如磷酸基)的化合物反应来引入放射性同位素卤素或放射性磷。
化合物或其药学上可接受的盐的具体实施方案如下:
(5-(((2-(二甲基氨基)乙基)氨基甲酰基)吡啶-2-基)氨基甲酸[131i]碘化物
n-(2-(二甲基氨基)乙基)-6-([125i]碘甲基)烟酰胺
n-(2-(二甲基氨基)乙基)-5-([125i]碘甲基)吡啶酰胺
n-(3-(二甲基氨基)丙基)-5-[131i]碘吡啶-2-甲酰胺
(s)-5-(2-氨基-3-(4-羟基-3-[131i]碘苯基)丙酰胺基)-n-(2-(二甲基氨基)乙基)吡啶甲酰胺
(s)-5-(4-(4-(2-(2-氨基-3-(4-羟基-3-[125i]碘苯基)丙酰胺基)乙基)-1h-1,2,3-三唑-1-基)丁酰胺基)-n-(2-(二甲基氨基)乙基)吡啶甲酰胺
(s)-6-(6-(3-(3-(2-氨基-3-(4-羟基-3-[125i]碘苯基)丙酰胺基)丙基)-3h-二苯并[b,f][1,2,3]三唑并[4,5-d]偶氮素-8(9h)-基)-6-氧己酰胺基)-n-(2-(二甲基氨基)乙基)烟酰胺
(6-((2-(二甲基氨基)乙基)氨基甲酰基)吡啶-3-基)甲基二氢[32p]磷酸酯)
4-乙酰氨基-n-(2-(二甲基氨基)乙基)-5-[131i]碘-2-甲氧基苯甲酰胺
177lu-dota-dmp
177lu-dota-dmpy2
177lu-dota-ncs-dmp
177lu-dota-ncs-dmpy2
177lu-dota-ncs-三唑-peg-dmp
177lu-dota-ncs-三唑-peg-dmpy2
177lu-dota-ncs-adibo-dmp
177lu-dota-ncs-adibo-dmpy2
177lu-dota-三唑-dmp
177lu-dota-三唑-dmpy2
177lu-dota-三唑-peg-dmp
177lu-dota-三唑-peg-dmpy2
177lu-dota-adibo-dmp
177lu-dota-adibo-dmpy2
式1至25中,dota是1,4,7,10-四氮杂环十二烷-1,4,7,10-四乙酸的缩写,ncs代表异氰酸酯,peg是聚乙二醇的缩写,dmp是4-氨基-n-(3-(二甲基氨基)丙基)苄酰胺的缩写,dmpy2是6-氨基-n-(3-(二甲基氨基)丙基)烟酰胺的缩写,而adibo为氮杂二苯并环辛炔。
另一方面,式2的化合物可以通过如下所示的方案2首先制备螯合剂结合化合物,然后向其中添加放射性同位素金属元素来制备:
如以上方案1中所示,可以使母体化合物与六甲基二锡(hexamethylditine)反应以用三甲基锡基团取代卤素原子,然后与包含放射性同位素的卤素化合物反应。此时,r1部分是放射性同位素部分,l1对应连接放射性同位素部分和黑素瘤靶向部分的接头,并且除r1和l1之外的右结构对应于黑素瘤靶向部分。同样,如方案2所示,可以通过与黑色素瘤靶向部分结合来制备螯合剂部分,并且作为螯合剂部分的反应性基团的异硫氰酸酯(ncs)或琥珀酰亚胺基团可分别通过硫脲键或酰胺键和黑色素瘤靶向部分的胺基连接,并且反应性基团和键合方法是示例性的,并且不限于方案2。
在方案1或2中,接头l1可以是将螯合剂通过点击化学连接至黑素瘤靶向部分的模块连接部,所述点击化学例如叠氮化物-炔烃环加成反应,通过除了一般有机合成的、点击化学的炔烃-亚硝基反应,(炔烃-硝基环加成)反应,烯烃和叠氮化物[3+2]环加成反应,烯烃和四嗪逆需求diels-alder)反应,以及除了一般有机合成的、炔烃和四嗪光点击反应形成三唑或二苯并三唑并恶唑啉(dibenzotriazoloazocine)。模块化接头可包括一个或多个功能性接头,包括烃链接头(-[ch2]n-)或聚乙二醇(-[c2h4o]n-),其用作放射性同位素部分与黑素瘤靶向部分之间隔开一定距离的间隔基,及疏水部分(例如,
在所述的新的放射性化合物或其药学上可接受的盐中,所述化学键可以是酯键,酰胺键,醚键,硫醚键,硫酯键或二硫键,并且l2可更优选为具有1至5个碳原子的亚烷基(-(ch2)n-,n为1至5的整数),最优选为亚乙基(-ch2ch2-)或亚丙基(-ch2ch2ch2-)。
方案2中所示的螯合剂是示例性的,并且可以使用上述各种螯合剂。
“药学上可接受的盐”优选为使用无机酸或有机酸的盐,更优选为使用脂肪族例如甲氧基、乙酰氧基、三氟乙酰氧基阴离子、氯盐、溴盐、碘盐、芳族的盐,或者可以使用诸如芳基脂族羧酸盐、硝酸盐、硫酸盐、磷酸盐、磺酸盐、甲磺酸盐、苯磺酸盐和甲苯磺酸盐,但不限于此。另外,本发明的药学上可接受的盐包括使用f-、cl-、br-或i-的盐。然而,本发明的药学上可接受的盐不限于此。
根据本发明的另一个方面,提供了一种用于治疗黑素瘤的药物组合物,其包含所述放射性化合物或其药学上可接受的盐作为活性成分。
在实际使用中,根据常规药物制备技术,根据本发明的一个实施方案的药物组合物可以与药学上可接受的载体组合。根据所需的制剂,例如口服或肠胃外施用(包括静脉内施用),载体可以采取多种形式。
另外,根据本发明的一个实施方案的药物组合物可以以0.1mg/kg至1g/kg,更优选0.1mg/kg至500mg/kg的剂量给药。另一方面,可以根据患者的年龄、性别和状况在每天或每年允许的辐射暴露范围内适当地调整剂量。
根据本发明的一个实施方案的药物组合物还包括惰性成分,所述惰性成分包括药学上可接受的载体。如本文所用,“药学上可接受的载体”是指除活性成分以外的组合物,特别是药物组合物的组分。这种药学上可接受的载体的例子包括粘合剂、崩解剂、稀释剂、填充剂、润滑剂、增溶剂或乳化剂和盐。
所述的新型药物组合物可以通过肠胃外施用给予受试者,并且肠胃外给药可以是静脉内注射,腹膜内注射、肌肉内注射或皮下给药,但是静脉内给药是最优选的。
在下文中,将通过实施例和实验实施例更详细地描述本发明。然而,本发明不限于下面公开的实施例,而是可以以各种不同的形式来实现,并且提供实施例是为了使本领域技术人员完全理解本发明的范围。
实施例1:前体的制备
1-1:dota-dmp的制备
将5g的4-氨基苯甲酸和0.987g的n,n,n',n'-四甲基-o-(n-琥珀酰亚胺基)脲四氟硼酸酯(tstu)溶解在n,n-二甲酰基甲酰胺(dmf)中,加入1.72ml的n,n-二异丙基乙胺(dipea),然后将混合物使用回流装置在60℃下搅拌3小时。搅拌3小时后,加入0.612ml的n,n-二甲基乙二胺(dmeda),然后在室温下搅拌2小时。用60ml的ch2cl2和130ml的h2o萃取产物,并用mgso4除去ch2cl2层的水分并过滤。将滤液旋干,并且使用柱色谱法分离并纯化产物4-氨基-n-(2-(二甲基氨基)乙基)苯甲酰胺。
随后,将以上制备的0.08g的4-氨基-n-(2-(二甲基-氨基)乙基)苯甲酰胺和0.229g的dota-nhs酯溶解在chcl3中,并使用三乙胺将ph调节至9-10,然后在室温下搅拌24小时。通过减压蒸馏除去溶剂后,将具有上式26结构的2,2',2″-(10-(2-(4-(2-(2-(二甲基氨基))乙基-氨基甲酰基)苯基氨基)-2-氧代乙基)-1,4,7,10-四氮杂环十二-烷-1,4,7-三基)三乙酸使用半制备柱分离出,即为“dota-dmp”。nmr数据如下:
1h-nmr(300mhz,d2o):2.92(s,6h),3.13(br,16h),3.33(t,2h),3.74(t,2h),3.87(br,8h),6.69(d,2h),7.66(d,2h).
1-2:制备dota-dmpy2
将0.6g的5-氨基吡啶-2-羧酸和1.308g的tstu溶解在dmf中,并添加2.41ml的dipea,然后使用回流装置在60℃下搅拌3小时。搅拌3小时后,加入0.808ml的dmeda,然后在室温下搅拌2小时。用80ml的ch2cl2和130ml的h2o萃取产物,并用mgso4除去ch2cl2层的水分并过滤。将滤出的滤液旋干,并分离出5-氨基-n-(2-(二甲基氨基)乙基)吡啶甲酸酰胺(nh2-dmpy2),并使用柱色谱法纯化。
随后,将以上制备的0.07g的nh2-dmpy2和0.2g的dota-nhs酯溶解在chcl3中,使用三乙胺将ph调节至9-10,然后在室温下搅拌24小时。随后,在减压下进行蒸馏以除去溶剂,然后将具有上述式27结构的2,2',2″-(10-(2-((6-(((2-(二甲基氨基)乙基))氨基甲酰基)吡啶-3-基)氨基)-2-氧乙基)-1,4,7,10-四氮杂环十二烷-1,4,7-三基)三乙酸使用半制备柱分离,即为“dota-dmpy2”。dota-dmpy2的1h-nmr分析结果如下:
1h-nmr(300mhz,d2o):2.99(s,6h),3.16(br,16h),3.41(t,2h),3.77(t,2h),3.85(br,8h),7.93(d,1h),8.35(m,1h),8.88(d,1h).
1-3:制备dota-nsc-dmp
将0.08g的实施例1-1中制备的nh2-dmfb和0.208g的2-(4-异硫氰酸根合苄基)-1,4,7,10-四氮杂环十二-烷-1,4,7,10-四乙酸(p-scn-bn-dota)溶解于chcl3中,并使用三乙胺将ph调节至9-10,然后在室温下搅拌24小时。通过减压蒸馏除去溶剂后,将2,2',2″,2″′-(2-(4-(3-(4-(((2-(二甲基氨基)乙基)氨基甲酰基)苯基)硫脲基)苄基)-1,4,7,10-四氮杂-环十二烷-1,4,7,10-四基)四乙酸用半制备柱分离,即为“dota-ncs-dmp”。dota-ncs-dmp的1h-nmr数据如下:
1h-nmr(300mhz,d2o):2.92(s,6h),3.13(br,16h),3.33(t,2h),3.74(t,2h),3.87(br,8h),6.43(d,2h),6.69(d,2h),6.86(d,2h),7.66(d,2h).
1-4:制备dota-ncs-dmpy2
将0.07g的实施例1-2中制备的nh2-dmpy2和0.181g的p-scn-bn-dota溶解在chcl3中,使用三乙胺将ph调节至9-10,然后在室温下搅拌24小时。通过减压蒸馏除去溶剂后,将2,2',2″,2″′-(2-(4-(3-(6-(((2-(二(甲基氨基)乙基)氨基甲酰基))吡啶-3-基)硫脲基)苄基)-1,4,7,10-四氮杂-环十二烷-1,4,7,10-四基)四乙酸用半制备柱分离,即为“dota-ncs-dmpy2″。dota-ncs-dmpy2的1h-nmr数据如下:
1h-nmr(300mhz,d2o):2.99(s,6h),3.16(br,16h),3.41(t,2h),3.77(t,2h),3.85(br,8h),6.42(d,2h),6.87(d,2h),7.93(d,1h),8.35(m,1h),8.88(d,1h).
实施例2:标准材料的制备
2-1:lu-dota-dmp的制备
将5mg的实施例1-1中制备的dota-dmp和5mg的lucl3溶解在0.2m乙酸钠缓冲液中,并在95℃下搅拌1小时。使用半制备柱分离具有上式30的结构的lu-dota-dmp。lu-dota-dmp的1h-nmr数据如下:
1h-nmr(300mhz,d2o):2.94(s,6h),3.12(br,16h),3.34(t,2h),3.72(t,2h),3.85(br,8h),6.68(d,2h),7.65(d,2h).
2-2:preparationoflu-dota-dmpy2
将7mg的实施例1-2中制备的dota-dmpy2和7mg的lucl3溶解在0.2m乙酸钠缓冲液中,并在95℃搅拌1小时。使用半制备柱分离具有上式31的结构的lu-dota-dmpy2。lu-dota-dmpy2的1h-nmr数据如下:
1h-nmr(300mhz,d2o):2.98(s,6h),3.14(br,16h),3.38(t,2h),3.79(t,2h),3.88(br,8h),7.91(d,1h),8.38(m,1h),8.90(d,1h).
2-3:制备lu-dota-ncs-dmp
将5mg的实施例1-3中制备的dota-ncs-dmp和4mg的lucl3溶解在0.2m乙酸钠缓冲液中,并在95℃下搅拌1小时。使用半制备柱分离具有以上式32的结构的lu-dota-ncs-dmp。lu-dota-ncs-dmp的1h-nmr数据如下:
1h-nmr(300mhz,d2o):2.94(s,6h),3.12(br,16h),3.34(t,2h),3.72(t,2h),3.85(br,8h),6.45(d,2h),6.68(d,2h),6.85(d,2h),7.65(d,2h).
2-4:制备lu-dota-ncs-dmpy2
将7mg的实施例1-4中制备的dota-ncs-dmpy2和6mg的lucl3溶解在0.2m乙酸钠缓冲溶液中,并在95℃搅拌1小时。将具有上式33的结构的lu-dota-ncs-dmpy2使用半制备柱分离。lu-dota-ncs-dmpy2的1h-nmr数据如下:
1h-nmr(300mhz,d2o):2.98(s,6h),3.14(br,16h),3.38(t,2h),3.79(t,2h),3.88(br,8h),6.43(d,2h),6.89(d,2h),7.91(d,1h),8.38(m,1h),8.90(d,1h).
实施例3:放射性化合物的制备
3-1:制备177lu-dota-dmp
将30μg的实施例1-1中制备的dota-dmp和177lucl3(20mci)溶解在0.2m的乙酸钠缓冲溶液中后,将反应混合物在90℃下孵育1小时以合成具有上式12的结构的177lu-dota-dmp。在室温下冷却反应混合物后,使用半制备柱分离并纯化放射性化合物。
3-2:177lu-dota-dmpy2的制备
将30μg的实施例1-2中制备的dota-dmpy2和177lucl3(30mci)溶解在0.2m乙酸钠缓冲溶液中后,将反应混合物在90℃下温育1小时以合成具有上式13的结构的177lu-dota-dmpy2。在室温下冷却反应混合物后,使用半制备柱分离并纯化放射性化合物。
如上所述,将使用放射性同位素钌177lu标记的177lu-dota-dmpy2使用以柠檬酸水溶液作为显影溶剂的放射性薄层色谱分离,以分析标记产率和放射化学纯度。如图1所示,出现在显影距离20-40mm之间的峰表示通过本发明的实施例合成的177lu-dota-dmpy2,而出现在显影距离85-95mm之间的峰表示未标记的游离177lu。计算峰面积的结果,证实了以上合成的177lu-dota-dmpy2的放射化学纯度>98%或以上。
3-3:177lu-dota-ncs-dmp的制备
将30μg的实施例1-3中制备的dota-ncs-dmp和177lucl3(25mci)溶解在0.2m的乙酸钠缓冲溶液中后,将反应混合物在90℃孵育1小时以合成具有上式14的结构的177lu-dota-ncs-dmp。在室温下冷却反应混合物之后,将177lu-dota-ncs-dmp使用半制备柱分离并纯化。
3-4:制备177lu-dota-ncs-dmpy2
将30μg的实施例1-4中制备的dota-ncs-dmpy2和177lucl3(23mci)溶解在0.2m的乙酸钠缓冲溶液中,然后依次将反应混合物在90℃孵育1小时以合成具有上式15结构的177lu-dota-ncs-dmpy2。在室温下冷却反应混合物后,将177lu-dota-ncs-dmpy2使用半制备柱分离并纯化。
实验实施例1:体内抗癌活性试验
本发明人将实施例3-2中制备的放射性化合物静脉内施用于黑素瘤小鼠模型,并测量了肿瘤体积和体重随时间的变化。
特别地,使用雄性balb/cnu/nu小鼠(6周龄),并在全南国立大学医学院和顺医院的设施中饲养。这项研究方案得到了全南国立大学医学院的机构动物护理和使用委员会(iacuc)的批准。
将1×106个b16f10细胞系(小鼠黑素瘤细胞系)的细胞接种在雄性balb/cnu/nu小鼠的右肩上。在肿瘤体积增加到大约100至150mm3并稳定后,将实施例3-2中合成的177lu-dota-dmpy2以90mbq或120mbq的放射剂量静脉内注射到实验动物中,对照组仅使用磷酸盐缓冲液(pbs)。在施用后的21天以3天的间隔记录动物的肿瘤体积(mm3)和体重,并在测量肿瘤组织的宽度和长度之后,使用以下计算公式计算肿瘤体积:
肿瘤体积(mm3)=(宽度)2×(长度)×0.5
仅在治疗开始时施用一次放射性化合物。实验完成后,处死实验动物,切除肿瘤组织并测量体重。
结果如图2所示,施用了根据本发明的实施例的177lu-dota-dmpy2的实验动物中,肿瘤的大小显著减小,并且特别地,当放射剂量加倍时,已证实肿瘤几乎不增长。
另外,为了检查试验药物是否具有副作用,试验动物的体重。如图3所示的测量实验动物的体重变化的结果,施用根据本发明的一个实施例的177lu-dota-ncs-dmpy2的动物与对照组相比,体重没有显著差异。
工业实用性
如上所述,根据本发明的实施方案的放射性化合物可非常有效地用于开发用于治疗黑素瘤的放射性药物。
上述实施例和实验实施例更详细地描述了本发明。然而,上述实施例和实验实施例旨在更充分地描述本发明,而对于本领域普通技术人员显而易见的是,本发明的真实范围不限于上述实施例和实验例。因此,本发明的实际保护范围如所附权利要求书中所确定。
- 还没有人留言评论。精彩留言会获得点赞!