一种生成表面簇的方法与流程
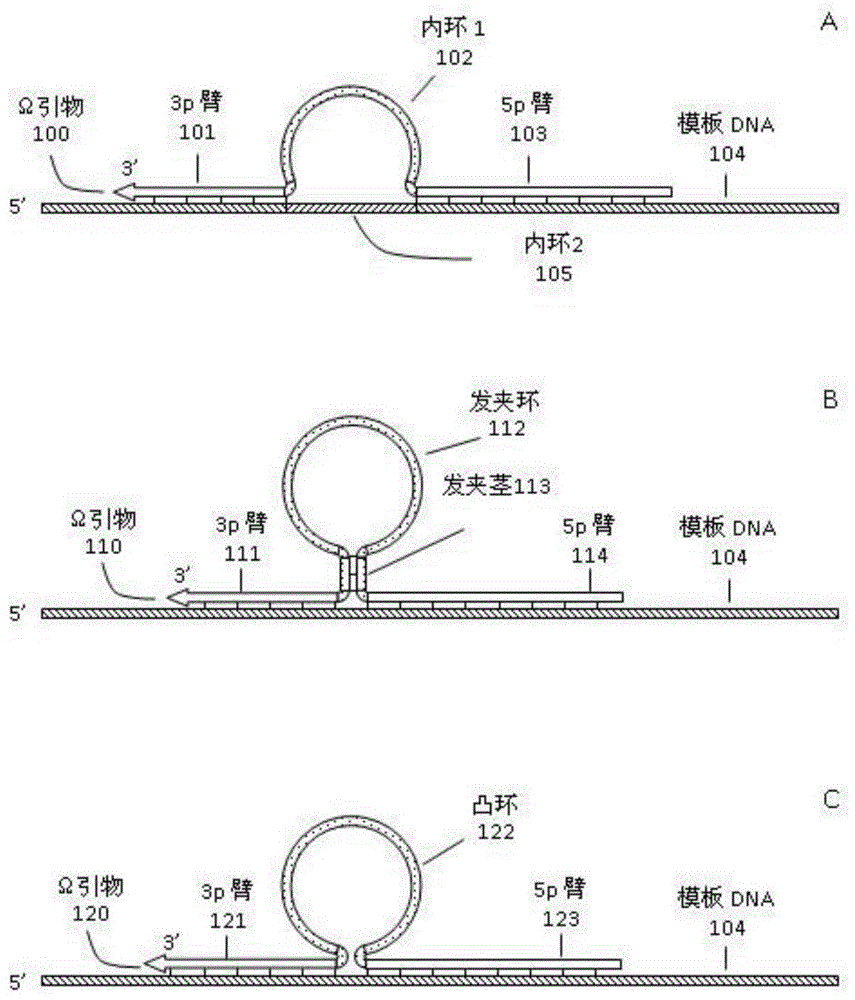
此申请是申请日2014年11月26日,申请号201480073756.7(国际申请号pct/us2014/067666),发明名称为“选择性扩增核酸序列”的分案申请。以前申请此申请要求2013年11月26日提交的美国序列号61/693,211的优先权,现将其全部纳入此申请中。发明领域本发明涉及核酸序列复制领域,包括聚合酶链反应(pcr)。具体而言,本发明涉及用于从一个或多个模板序列中扩增一个或多个靶序列的方法和组合物。特别地,本发明提供了新的引物设计来增强pcr反应的特异性。本发明还提供了方法和组合物,使得在单个反应管中能够使用特异性引物进行特定序列区段的选择并使用一对通用引物扩增所有选择的序列区段。背景许多生物和生物医学领域的应用,如遗传疾病筛查、个体医学、法医检验和靶向测序,需要从大量的核酸序列(如各种活细胞基因组dna,宏基因组样品核酸序列的混合物,肠道菌群的微生物组dna)中复制和(或)扩增一个或多个选定的核酸序列区段。pcr是一种从核酸混合物中选择性扩增目的核酸序列的强大工具。涉及多个目的序列的多重pcr应用已得到广泛应用。1988年,chamberlain等人在“deletionscreeningoftheduchennemusculardystrophylocusviamultiplexdnaamplification”这篇文章中首次报道了多重pcr法,文章发表在《nucleicacidsresearch》上(nucleicacidsres.1988,16:11141-11156)。但是,henegariu等人在1997年发表在《biotechniques》上的“multiplexpcr:criticalparametersandstep-by-stepprotocol”中描述道:由于繁重的优化工作要求,通常认为传统的多重pcr很难在每个反应中进行几十个以上的靶(biotechniques.1997,23:504-511)。正如edwards等人在1994年发表在《genomeresearch》上的“multiplexpcr:advantages,development,andapplications”中所述,多重pcr的主要挑战包括引物二聚体的形成、靶扩增的不均一性和错配率高(genomeres.1994,3:s65–75)。为了统一引物依赖性的pcr条件,a.p.shuber等人于1995年发表在《genomeresearch》上的“asimplifiedprocedurefordevelopingmultiplexpcrs”中描述了多重pcr的一个变体(genomeres.1995,5:488-493)。在多重pcr中使用了嵌合引物,每个嵌合引物都包含一个与序列特异性识别位点互补的3'区和由非相关的20个核苷酸序列组成的5'区。证明了包含嵌合基序的pcr引物对都使用了相同的反应条件、循环次数和退火温度。这种方法被认为有助于消除研发的多重pcr中所涉及的多个优化步骤。然而,单个引物浓度的调整仍然是必需的。本发明提供了一种消除单个引物浓度调整要求的解决方案。为降低引物二聚体的形成和减少错配事件的发生,多重pcr的变体已有报道。z.lin等人于1996年发表在《proceedingsofthenationalacademyofsciences》上的“multiplexgenotypedeterminationatalargenumberofgeneloci”中描述了一种转换多重扩增为单重扩增的方法,以减少引物与引物之间的相互作用(natl.acad.sci.1996,93:2582-2587)。实施方法的过程包括三轮独立的pcr。在前两轮pcr中,位点特异引物,含有5'端通用尾部序列(tail)或标记序列(tag),用于将通用尾部序列添加进靶序列。然后,在第3轮中,所有通用的尾部标记的靶(26个基因位点)可被与通用尾部序列匹配的一对通用引物同时地扩增。第一轮和第二轮类似于传统的多重pcr,需要一个耗时的涉及引物浓度调整的优化过程。此外,工作流程含有多个人工操作步骤,包括三轮独立的pcr和pcr轮之间pcr产物的纯化。作为比较,本发明提供了一种显著简化的工作流程。1991年,a.j.jeffreys等人发表在《nature》上的“minisatelliterepeatcodingasadigitalapproachtodnatyping”中描述了一种dna分型方法,称为小卫星变体重复作图pcr(mvr-pcr)(nature.1991,354:204-209)。每个pcr反应包括三种引物:一种10~20nm低浓度的5’标记的变体重复特异性引物;一种1μm高浓度的小卫星侧翼引物;一种1μm高浓度的标记引物,这个标记引物具有与变体重复特异性引物的标记区段匹配的序列。侧翼引物具有与小卫星区的侧翼区段匹配的序列。使用amplitaq(perkin-elmer-centus)的重组taq聚合酶。pcr反应循环为在dna热循环仪上,96℃变性1.3min,68℃退火1min,70℃延伸5min,共18个循环;随后,追加67℃退火1min,70℃延伸10min,共2个循环。一个反应可生成超过50个不同长度的扩增子,每个扩增长度表示特异变体重复单元和侧翼位点之间的距离。扩增子产生开始于一个热循环中的退火阶段,此时变体重复序列特异性引物退火结合dna样品中的小卫星区的匹配变体重复单元。接着,退火后的特异性引物延伸超出侧翼位点,通过聚合酶延伸反应形成第一个延伸产物。在下一个热循环中,侧翼引物退火结合至第一个延伸产物,并延伸产生末端与标记序列互补的第二个延伸产物。从下一个热循环开始,对于第二个延伸产物,高浓度的侧翼引物和标记引物配成一对,高效地生成pcr产物。有时候,由特异性引物的非pcr产物的内部引发会生成较短的真实pcr产物。这是第一个证明标记驱动(tag-driven)pcr可行性的工作,包含了,相对于标记的特异性引物,更高浓度的标记引物。另一方面,设计上述描述的方法是为了揭示共享一个靶特异性引物的重复序列单元模式。在每一个pcr反应混合物中总共只使用了三种引物。这项工作并没有提供如何处理通用类型多重pcr中涉及多对靶向特异性引物以及最小化引物二聚体形成的任何明显的解决办法。1997年,j.brownie等人发表在《nucleicacidsresearch》上的“theeliminationofprimer-dimeraccumulationinpcr”中描述了同源标记辅助的非二聚体系统(homo-tagassistednon-dimersystem,hands),一种为了减少引物二聚体的形成而设计的实验方法(nucleicacidsres.1997,25:3235-3241)。多个标记的基因组特异性引物采用低浓度,单一标记引物或通用引物采用高浓度。类似于上述jeffreys的方法,标记引物与标记的基因组特异性引物的5'尾部部分具有相同的序列。文章作者建议设计基因组特异性引物,如标记退火结合其互补序列的tm(熔融温度)应高于基因组特异性引发的双链体的tm。这种设计会使低退火温度下基因组特异性引物的基因组特异性部分的基因组引发转换为升高的退火温度下标记引导的尾部引发。基因组引发的两个循环后,标记序列的互补序列被掺入扩增子末端。然后退火温度升高和随后的扩增很大程度上由标记引物驱动。所有产生的扩增子都具有相同的一对互补末端。当扩增子很短,只有100至120个核苷酸,像引物二聚体一样,互补末端趋向于引起发夹结构。这些发夹结构的形成阻止了其他标记引物的退火结合,因而阻止了非特异性引物二聚体产物的积累。hands方法的缺点是短靶序列产物产率降低。通过设计,该方法抑制短序列的扩增,无论是不需要的引物二聚体或需要的靶序列。事实上,报告的数据揭示,当靶产物的长度从550减少到300个核苷酸,其产物产率显著降低的模式。这使得该方法不适用于涉及靶扩增子长度小于300个核苷酸的应用。这样的应用包括用于高度并行测序的序列富集。上面描述的jeffreys和brownie的这两种方法都使用了taq聚合酶。taq聚合酶具有人所共知的5'-3'核酸内切酶的活性和5'flap核酸内切酶的活性。p.m.holland等(1991年)和v.lyamichev等(1993年)等人分别发表在《proceedingsofthenationalacademyofsciences》和《sciences》上的“detectionofspecificpolymerasechainreactionproductbyutilizingthe5'-3'exonucleaseactivityofthermusaquaticusdnapolymerase”和“structure-specificendonucleolyticcleavageofnucleicacidsbyeubacterialdnapolymerases”中有详细的介绍(proc.natl.acad.sci.1991,88:7276-7280;science.1993,260:778-783)。当延伸的dna片段遇到聚合酶,这些核酸酶活性会降解双链dna。这就对在两个或多个靶区域是串联或位置接近的应用具有局限性。与串联区域的中间杂交的引物很有可能被降解,且未能产生预期的扩增子。这样的现象确实出现在上述jeffreys文章数据中。本项发明的一个方面通过使用新的缺乏5'-3'外切核酸酶活性和链置换活性的聚合酶克服了上述限制。另一个用于多重扩增应用的标记驱动pcr方法由b.frey等人于2013年在标题为“methodsandamplificationoftargetnucleicacidsusingamulti-primerapproach”的美国专利申请书(us2013/00045894a1)中公开。与jeffreys和brownie的方法类似,在一个扩增反应中包含两组引物(标记的靶特异性引物和通用引物)用于扩增靶核酸。一个不同的反应条件的特征是建议特异性引物组浓度与通用引物组相同或比其更高。因此,在可比较的浓度水平,反应产物包含侧翼有标记的特异性引物的较短序列和侧翼有通用引物的较长序列。作者建议在进行相应的应用前,将较长的序列(所需的产物)从较短的序列(不想要的产物)中分离出来。本项发明识别了显著不同的反应条件,其产生的纯化产物绝大部分都是预期的全长序列。高度多重核酸检测技术,如微阵列和大规模平行测序技术的进步已经使得在一次检测运行中分析成百上千、数以百万计的核酸序列成为可能。这些检测技术的成功应用,往往有一个共同的特点,即有一个要求,在实际检测前需要扩增目的区域。在不同的文章中描述了采用表面固定的引物对进行高度多重扩增的方法,如a.pemov等人2005年在《nucleicacidsresearch》上发表的“dnaanalysiswithmultiplexmicroarray-enhancedpcr”和l.s.meuzelaar等人2007年在《naturemethods》上发表的“megaplexpcr:astrategyformultiplexamplification”以及其他的参考文献中(nucleicacidsres.2005,33:e11;nat.methods.2007,4:835-837)。多个靶特异性引物对通过它们的5'末端固定在固体表面或凝胶基质内。固定化由于引物的物理分离被认为有助于避免引物二聚体的形成。每个引物包括3’侧靶特异性启动部分和5’侧通用启动部分。固定化的引物被用来选择性地复制靶序列,并在固相pcr过程中将通用启动部分掺入到复制序列中。然后,一对通用引物被用于扩增固相pcr产物。使用单一的通用引物对能消除靶特异性引物扩增的偏差。但是,固相pcr的使用使得操作过程复杂,有待于研究出一种更简单的方法。2008年,k.e.varley等人在《genomeresearch》上发表的“nestedpatchpcrenableshighlymultiplexedmutationdiscoveryincandidategenes”中描述了使用可切割引物的液相多重pcr(genomeres.2008,18:1844-1850)。该方法依赖于两轮靶特异性的富集。首先,针对每个靶设计引物对,并将引物混合物运用到预先确定循环数的多重pcr中。引物含有代替胸腺嘧啶的尿嘧啶碱基,这样扩增后,在尿嘧啶dna糖基化酶,核酸内切酶viii和外切核酸酶i的作用下,可以有效地将引物区域从扩增子中移除。在第二轮选择中,运用了嵌套补丁衔接头(nestedpatchadaptors)。这些衔接头包括双链通用区段和靶特异性的单链突出端。嵌套补丁衔接头与引物殆尽的扩增子进行杂交和连接之后,使用对应通用序列的引物进行多模板pcr扩增。因为这种连接依赖于限制性多重pcr中所用的原始引物内部紧邻的序列,所以嵌套补丁衔接头增强了特异性。j.leamon等人于2012年在标题为“methodsandcompositionsformultiplexpcr”的美国专利申请书(us2012/0295819a1)中描述了这种方法的一种变体。这种方法通过平性末端连接通用衔接头省去了第二轮选择。该方法的这两个版本有着相似的工作流程,都需要多个人工操作步骤。此外,在第一轮多重pcr中,每个靶的扩增都需要在每个靶特异性引物下进行。不可避免地,随着热循环次数的增加,引物相关的产率差异会呈指数放大,这会导致靶扩增的不均一性。本发明的一个方面是提供一个简单的工作流程,并在液相中实现了均一的高多重扩增。附图说明图1:所公开的ω引物的三个示例性设计的示意图。a.含有内环的ω引物;b.含有发夹环的ω引物;c.含有凸环的ω引物。图2:所公开的ω探针的三个示例性设计的示意图。图3:运用常规特异性引物的接力pcr方法示范性实施例的示意性轮廓。图4a:运用含有内环的ω特异性引物的接力pcr方法示范性实施例的示意性轮廓。图4b:运用含有凸环的ω特异性引物的接力pcr方法示范性实施例的示意性轮廓。图5:运用含有多环的ω特异性引物的接力pcr方法示范性实施例的示意性轮廓。图6:在靶选择中运用单一ω特性引物的单个引物延伸接力pcr方法示范性实施例的示意性轮廓。图7:本发明纯化方法的示范性实施例的示意图。图8:本发明靶固定方法的示范性实施例的示意图。图9:涉及前体扩增和激活的特异性引物产生的示范性实施例的示意图。图10:涉及pcr扩增和du消化的帽子移除的特异性引物产生的示范性实施例的示意图。图11:涉及pcr扩增和限制性酶切消化的帽子移除的特异性引物产生的示范性实施例的示意图。图12:引物设计计算流程图。图13:基因组区域变体等位基因装配示意图。图14:同时分子内折叠和分子间杂交的平衡反应示意图。图15:涉及ω引物和模板之间结合的同时链内折叠和链间杂交的平衡反应示意图。图16:λdna样本中常规pcr和接力pcr产物的琼脂糖凝胶电泳图举例。泳道1:常规pcr,特异性引物1和2=500nm,λdna=10fm;泳道2:接力pcr,特异性引物1和2=5nm,通用引物1和2=500ηm,λdna=10fm;泳道3:接力pcr,特异性引物1和2=0.5nm,通用引物1和2=500nm,λdna=10fm;泳道l:显示对应标准参照物大小(碱基对或bp)的dna序列梯的走胶。图17:人类基因组dna样品中使用ω引物的接力pcr的琼脂糖凝胶电泳图举例。泳道1到泳道6:6个单独pcr的产物的走胶,其中每个pcr使用了针对单一靶区域的一对ω引物,ω引物显示在试验ii相应表格中。这6个单独pcr走胶中也使用了相同的通用引物对;泳道7:非特异性引物对照走胶的结果;泳道l:显示对应标准参照物大小(碱基对或bp)的dna序列梯。图18:使用ω引物的多重接力pcr的试验结果。a.人类基因组dna样品中使用ω引物的多重接力pcr的琼脂糖凝胶电泳图举例。泳道1:多重接力pcr产物的走胶,包含了图5单个接力pcr中使用的6对ω引物和一对通用引物;泳道l:显示对应标准参照物大小(碱基对或bp)的dna序列梯。b.6个预期扩增子测序读数分布的散布图。图19:多重pcr产物中扩增子读数分布的高通量测序测量结果。图a、b和c分别是使用浓度为每个引物1nm、0.2nm和0.04nm的特异性ω引物得到的。图20:使用来自微阵列合成寡核苷酸的ω引物的多重接力pcr实验结果。a.微阵列合成引物前体模板的pcr产物琼脂糖凝胶电泳图。泳道1:含有204个寡核苷酸序列的混合模板库的pcr产物;泳道l:显示对应标准参照物大小(碱基对或bp)的dna序列梯。b.人类基因组dna样品使用来自图20a的引物前体混合物的微阵列的多重接力pcr产物琼脂糖凝胶电泳图。泳道1:多重pcr走胶的产物;泳道l:显示对应标准参照物大小(碱基对或bp)的dna序列梯。图21:特异性引物的酶法制备及其在多重接力pcr中特异性引物使用的实验结果。a:特异性引物模板的pcr产物琼脂糖凝胶电泳图。泳道1:混合模板的pcr产物;泳道l:显示对应标准参照物大小长度(碱基对或bp)的dna序列梯。b:泳道2显示的是限制性酶切消化产物的凝胶图。泳道1:限制性酶切消化之前的原始pcr产物。泳道l:显示对应标准参照物大小(碱基对或bp)的dna序列梯。c:接力pcr产物图。泳道1:来自化学合成特异性引物的阳性对照结果;泳道2:来自酶法制备的特异性引物的接力pcr产物;泳道3:阴性对照结果;泳道l:显示对应标准参照物大小(碱基对或bp)的dna序列梯。定义术语“管”是指一种发生pcr或任何其它类型生物分子反应的容器,这种“管”可以由塑料制成,并是微管或eppendorf管的形式。这种“管”也可以由玻璃、硅酮、硅和金属制成,且是微加工装置的一部分。术语“靶”、“靶序列”和它们的衍生词,指被怀疑或预计将存在于样品中,并被设计以使用任何合适的方法被选择、分析、检测、探测、捕获、复制、合成和(或)扩增的单链或双链核酸序列。术语“文库”、“dna文库”和它们的衍生词,如用于此处,指的是dna片段或dna序列的集合,它们受到平行的样品制备和(或)平行检测测定过程。在一些实施例中,如多重扩增,使用靶特异性引物选择性扩增靶序列,并形成靶序列文库。术语“样品”指的被怀疑包含靶的任何样本、培养物等。样品可包括含有一种或多种核酸的任何生物、临床、外科、农业、大气或水生型样本。样品可包括任何类型的样本,如颊组织、全血、干燥血点、器官组织、血浆、尿、粪便、皮肤和毛发。该术语还包括任何分离的核酸样品,如新鲜冷冻或福尔马林固定的石蜡包埋组织的基因组dna。术语“合成”、“合成的”和它们的衍生词通常指的是聚合酶引导的核苷酸聚合反应,可选地以模板依赖方式。聚合酶通过将核苷三磷酸(ntp)、脱氧核苷三磷酸(dntp)或双脱氧核苷三磷酸(ddntp)中的核苷单磷酸转移到延伸的寡核苷酸链的3'羟基上,以合成寡核苷酸。为达到本次公开的目的,合成包括通过从脱氧核苷三磷酸转移核苷单磷酸的方式,杂交引物的连续延伸,。术语“寡”、“寡核苷酸”和它们的衍生词,是指短的单链核酸序列,包括dna、rna、dna-rna杂交体和包含分子的各种修饰基团。可以通过化学方法、酶促方法或者化学和酶促方法的组合来产生或合成它们。术语“延伸”及其变体,如用于此处,是指在给定引物和具有体内或体外酶促活动特性的给定聚合酶引导下,一个或多个核苷酸聚合到现有核酸分子的末端。典型地但不必要的,这样的引物延伸发生在模板依赖性方式中。模板依赖性延伸期间,碱基的顺序和选择是由既定的碱基配对规则驱动,包括沃森-克里克型(watson-crick)类型的碱基配对规则或替代地(特别是在涉及核苷酸类似物延伸反应的情况下)一些其它类型的碱基配对范例。在一个非限制性的例子中,在聚合酶的作用下,延伸发生在核酸分子3'oh端的核苷酸聚合反应中。术语“扩增”和“扩增的””和它们的衍生词通常指,核酸分子(被称为模板核酸分子)中至少有一部分被复制或拷贝,产生至少额外的核酸分子的行动或过程。额外的核酸分子选择性地包含与模板核酸分子至少一些部分基本上相同或基本上互补的序列。模板核酸分子可以是单链或双链,额外的核酸分子也可以独立地是单链或双链。在某些实施例中,扩增包括模板依赖性体外酶催化反应,用于产生核酸分子的至少一些部分的至少一个拷贝或者产生与核酸分子的至少一些部分互补的核酸序列的至少一个拷贝。扩增可选地包括核酸分子线性或指数的复制。在一些实施例中,这种扩增是在等温条件下进行的;在另一些实施例中,这种扩增可以包括热循环。在一些实施例中,扩增是一种多重扩增,单个扩增反应中包括多个靶序列的同时扩增。在单个扩增反应中,至少有一些靶序列位于相同核酸分子上或不同靶核酸分子中。在一些实施例中,“扩增”包括至少部分dna和rna碱基的单独扩增,或者组合扩增。扩增反应可包括单链或双链核酸底物,还可以进一步包括本领域技术人员已知的任何扩增过程。在一些实施例中,扩增反应包括聚合酶链反应(pcr)。术语“引物”和其派生词是指可以与目的靶序列杂交的任何多核苷酸或寡核苷酸。在本次公开的某些实施例中,至少引物的3’末端部分与靶序列的一部分互补。通常情况下,在扩增或合成条件下,引物作为扩增或合成的起始点,这样的扩增或合成可发生在模板依赖的方式中,并可选地形成引物延伸产物,该产物与靶序列的至少一部分互补。示例的扩增或合成条件可以包括引物与多核苷酸模板(例如,包括靶序列的模板)接触,核苷酸和诱导剂如在合适温度的聚合酶,盐浓度以及ph值,诱导核苷酸聚合到靶特异性引物的末端。扩增或合成反应中,引物可以与相配的引物配对,以形成包括正向引物和反向引物的引物对。在某些实施例中,正向引物包括与核酸分子链的至少一部分基本上互补的3’部分,反向引物包括与核酸分子链的至少的一部分基本上相同的3’部分。在一些实施例中,正向引物和反向引物能够杂交核酸双链体的相反链。可选地,正向引物启动第一条核酸链的合成,反向引物启动第二条核酸链,其中第一和第二条链彼此基本互补,或可杂交形成双链核酸分子。术语“特异性引物”和“靶特异性引物”是指单链寡核苷酸,它的3’部分与含有靶序列的核酸分子的至少一部分基本互补或相同。在某些实施例中,特异性引物包括两个或更多与含有靶序列的核酸分子的一部分基本互补或相同的特异性部分。在某些实施例中,该特异性引物包括至少通用区段。通用区段是设计为多对引物共享的序列片段。在某些实施例中,通用区段位于两个特异性部分之间。在某些实施例中,通用区段位于特异性引物的5’末端部分。术语“通用引物”和“文库引物”指单链寡核苷酸,其包含与核酸分子的通用区段的至少一部分基本互补或相同的3’部分。在一些实施例中,核酸分子的通用区段是在靶序列的pcr产物中。在一些实施例中,核酸分子的通用区段是在靶序列的引物延伸产物中。术语“杂交”与本领域的使用一致,一般指两个核酸分子经历碱基配对的相互作用的过程。两个核酸分子杂交时,一个核酸分子的任何部分是与其他核酸分子的任何部分碱基互补;它不一定要求两核酸分子在其整个各自的长度杂交。在一些实施例中,在核酸分子中至少一个可以包括不与其他核酸分子杂交的部分。术语“聚合酶链式反应”或“pcr”是k.b.mullis美国专利编号4683195和4683202中的一种方法,由此并入其为参考文献,其描述了增加未克隆纯化的基因组dna中目的多核苷酸片段的浓度的方法。扩增目的多核苷酸的这一过程包括向包含期望的目的多核苷酸的dna混合引入大量过量的两种寡核苷酸引物,随后在dna聚合酶的存在下,进行精确序列的热循环。两种引物与它们各自的目的双链多核苷酸链互补。为了实现扩增,将混合物变性、引物退火,与目的分子的多核苷酸内它们的互补序列结合。在退火后,引物在聚合酶下延伸,形成新的一对互补链。变性、引物退火和聚合酶延伸这三个步骤可多次重复(即变性,退火和延伸构成一个循环,可以有大量的“循环”)以获得高浓度预期的目的多核苷酸的扩增片段。预期的目的多核苷酸扩增片段(扩增子)的长度是由彼此相对的该对引物的相对位置确定的,因此,这个长度是一个可控参数。通过重复该过程,该方法被称为“聚合酶链式反应”(以下简称“pcr”)。因为混合物中目的多核苷酸的预期扩增片段成为占主导地位的核酸序列(从浓度方面),它们被称为“pcr扩增”。术语“单链pcr”和“线性扩增反应”指的是只使用每个引物特异性序列组中的一个引物进行的扩增反应。“引物特异性序列”是指含有至少一个引物互补部分的核酸序列。引物特异性序列组包含一个或多个引物特异性序列。术语“聚合酶”和它的衍生词一般是指能催化核苷酸聚合成核酸链的任何酶。通用但不是必须的,这种核苷酸聚合可以通过在模板依赖性方式发生。这样的聚合酶可包括但不限于天然存在的聚合酶和任何亚基及其截短,突变体聚合酶,变体聚合酶,重组体,融合或以其他方式基因改造的聚合酶,化学改性聚合酶,合成分子或装配体,以及任何类似物,衍生物或其保留了催化这样的聚合能力的片段。可选地,聚合酶可以是包括一个或多个突变的突变聚合酶,突变涉及与其他氨基酸的一个或多个氨基酸置换,聚合物的一个或多个氨基酸的插入或缺失,或者两个或多个聚合酶部分的连锁。一些示例性聚合酶包括但不限于dna聚合酶和rna聚合酶。这里所使用的术语“聚合酶”及其变体,也涉及包括彼此连接的至少2个部分的融合蛋白,其中第一部分包括能够催化核苷酸聚合成核酸链的肽,并与含有第二多肽的第二部分相连。在一些实施例中,该第二多肽可包括报道酶或持续合成能力增强域。可选地,聚合酶具有5’外切核酸酶活性或末端转移酶活性。在一些实施例中,聚合酶可以选择性的再激活,例如通过使用热、化学物质或额外再加入新的大量的聚合酶到反应混合物中。在一些实施例中,聚合酶可以包括热稳定,热启动,保真度高,3’至5’的核酸酶活性,5’至3’的核酸酶活性,和链置换活性。术语“多重扩增”,“多重pcr”及它们的衍生词术语是指使用至少一个靶特异性引物的样品中进行的两个或多个靶序列的选择性和非随机扩增。在一些实施方案中,多重扩增使得一些或所有靶序列在单个反应容器内扩增。在一个给定的多重扩增中,“多重”或通常指的是该单个多重扩增过程中扩增的不同的靶特异性序列的数量。术语“gc含量”指的是核酸分子的胞嘧啶和鸟嘌呤的含量。术语“dna条线码”及它的衍生词,通常指的是通用引物中单一短(4-14核苷酸)的核酸序列,可以作为区分或分离样品中多个扩增靶序列的“钥匙”。术语“互补的”和“互补”和它们的变体是指任何两个或多个核酸序列(例如,部分或整体模板核酸分子,靶序列和/或引物),可以逆平行在两个或多个对应位置进行碱基配对累积,如在杂交双链体中。这样的碱基配对可以根据一套既定的规则,如根据watson-crick碱基配对的规则或根据一些其他碱基配对模式。可选地,第一和第二核酸序列之间可以存在“完全”互补性,其中在第一核酸序列的每个核苷酸可以与第二核酸序列上对应的逆平行位置中的核苷酸进行稳定的碱基配对的相互作用。“部分的”互补性描述了核酸序列,其中一个核酸序列的残基的至少20%,但是少于100%与其他核酸序列的残基互补。“部分的”互补性也描述了核酸序列,其中一个核酸序列的目的部分的残基的至少20%,但是少于100%与其他核酸序列的残基互补。术语“缔合部分”、“结合系数”、“结合部分”和它们的衍生词指的是与相应引物杂交的模板部分、与相应探针杂交的靶或与相同的核酸序列的另一个部分杂交的部分。缔合部分的计算于2005年在miura等的“anovelstrategytodesignhighlyspecificpcrprimersbasedonthestabilityanduniquenessof3’-endsubsequences”(bioinformatics214363-4370)中有描述,且在本发明的说明中也有描述。术语“平行测序”、“大量平行测序”、“高通量测序”、“下一代测序”和它们的变体指的是并行测序过程中,同时产生数千或数百万条序列的测序技术。这项技术中牵涉到的各种方法和过程在2008年mardiser的"next-generationdnasequencingmethods"(annurevgenomicshumgenet9:387–402)和2010年m.l.metzkerx的“sequencingtechnologies—thenextgeneration”(naturereviewgenetics11:31-46)中均有描述。术语“引发区域”、“引发部分”和它们的衍生词指的是目的靶序列的一部分,设计其是要与相应的引物基本上杂交。术语“探针序列”、“探针”和它们的衍生词是指为了达到检测、捕获和富集的目的而设计的与目的靶序列杂交的核酸序列。在一些实施例中,探针包含一个或多个荧光染料基团。在一些实施例中,探针包含一个或多个荧光淬灭基团。在一些实施例中,探针包含一个或多个供体和(或)受体荧光基团。在一些实施例中,探针序列是溶液中的自由分子。在一些实施例中,探针序列固定在珠表面上。在一些实施例中,探针序列固定在一个基本平坦的表面上。在一些实施例中,探针序列嵌入在凝胶中。术语“特异性”通常指的是在所有报道的事件中正确报道事件的部分。在一些实施例中,该术语指的是在引物延伸反应中所有引物已经延伸条件下的所有引物中目标靶的正确延伸的的引物部分。在一些实施例中,该术语指的是在pcr反应中所产生的所有pcr产物中正确pcr产物的部分。在一些实施例中,该术语指的是在杂交反应中的所有探针-靶对中完全匹配的探针-靶对的部分。术语“变体”、“序列变异”和它们的衍生词是指目的样本的序列部分,它与参考样品或参考序列中相同位置上的序列部分不同。变异包括点变异和结构变异。对于本公开的目的,该变异可以是与疾病相关的或非疾病相关的。点变异包括单核苷酸多态性或snp。结构变异包括短插入、短缺失、大的插入、大缺失、插入缺失(缺失后跟一个插入)、置换、重复、倒置、易位或任何其他类型的变异。术语“捕获区域”和它们的衍生词是指夹在两对引物之间,包括引物的靶特异性部分的靶序列区域。概述本发明的目的是提供一种新的、改进的且操作简单的pcr方法,该方法提高了序列特异性,具有低的引物相关的扩增偏差,并适合于多重扩增用途。本发明的一个应用方面涉及大量并行测序应用的靶富集的领域。目的靶序列是从基因组样品中选择,扩增,并在侧翼具有包含可选的dna条形码的测序引发部分。本发明的另一个应用方面涉及用于dna阵列基因分型应用的靶富集领域。目的靶序列是从基因组样品中选择,扩增,并用荧光染料或用缀合的配体进行标记。这两个应用中的靶富集方法中的技术方法有许多相似之处。e.h.turner和memanova等人分别于2009和2010年在“methodsforgenomicpartitioning”(annu.rev.genomicshum.genet.2009,10:263–284)和“target-enrichmentstrategiesfornextgenerationsequencing”(nat.methods.2010,7,111–118)中提供了该领域的详细技术综述。主要方法包括多重pcr,环化捕获和杂交捕获。该方法的性能是通过捕获的特异性、均一性、多重性、输入要求、可扩展性、流程简单和成本进行评估。本发明提供了具有显著改善性能的靶富集的方法和组合物。本发明涉及寡核苷酸引物,它包括具有3’端和5’端的3p臂、环部分以及具有3’端和5’端的5p臂。其中,5p臂与dna模板杂交,3p臂在与dna模板杂交的同时,提供聚合酶延伸的序列特异性,环部分位于5p臂和3p臂之间,不与dna模板结合。本发明涉及包括设计的寡核苷酸和靶核酸的稳定杂交结构。其中设计的序列和靶序列是稳定的,其中所述的杂交结构具有一个或多个单链环以及两个或多个双链体区段,其中每个环位于双链体区段之间。本发明涉及扩增靶核酸的方法,包括提供第一个特异性引物,第二个特异引物,第一通用引物,第二通用引物,靶核酸,聚合酶和核苷酸,进行靶筛选,该靶筛选包括两轮第一热循环程序,该第一热循环程序包括变性步骤,退火步骤和延伸步骤和进行扩增,该扩增包括两轮或多轮第二热循环程序,该第二热循环程序包括变性步骤,退火步骤和延伸步骤,从而扩增靶核酸。本发明涉及扩增两种或多种不同的靶核酸的方法,包括提供第一组特异性引物,包含两个特异性引物,其中每个引物特异性设计用于第一个靶核酸;第二组特异性引物,其中每个引物特异性设计用于第二个靶核酸,第一通用引物,第二通用引物和靶核酸。进行两轮第一热循环程序,该第一热循环程序包括变性步骤,退火步骤和延伸步骤);以及进行两轮或多轮第二热循环程序,该第二热循环程序包括变性步骤,退火步骤和延伸步骤),从而扩增靶核酸。本发明涉及纯化pcr产物的方法,包括添加pcr反应成分的混合物添加,该混合物包括第一通用引物和第二通用引物,dna片段、聚合酶、pcr缓冲液,其中靶序列侧翼有引发区段,其与第一和第二通用引物相同或互补,其中片段不包含引发区域,其中第二通用引物包括引发区段、修饰基因区段和标记区段探针接枝珠,其中探针有序列与标记区段的序列基本上互补,并经由杂交,通过与珠721促进pcr产物的捕获。本发明涉及序列文库的制备方法,包括用第一通用引物和第二通用引物扩增靶序列,每个通用引物包括引发区段,修饰基因区段和标记区段,用于产生含有单链标记的pcr产物,这个单链标记将引导液应用于基底,使得基底与探针杂交,从而在基底表面产生引导/探针对,洗去多余的引导液,将含pcr产物的单链标记加入到基底中,从而使单链标记、引导(guide)和探针在基底表面共杂交。详细描述靶特异性引物本发明的一个方面涉及一种引物形式,被称为ω引物。图1是ω引物100、110、120的三个典型结构设计的示意图。每个引物包括三个功能部分,包括3p臂101、111和121;环102、112和122;和5p臂103、113和123。3p臂101、111和121的功能之一是与模板dna104杂交,并提供聚合酶延伸反应的起点。5p臂103、113和123稳定ω引物与模板dna之间的结合。环102、112和122使得两臂分开。图1a示意性地表示出了具有内环102和105的ω引物100与。内环是双链3p臂101和5p臂103部分之间的引物100和模板104两者上的单链核酸序列部分。图1b示意性地表示出了带有发夹环的ω引物110。发夹环由引物序列形成,包括单链的环112和双链茎113。图1c示意性地表示出了带有凸环122的ω引物120。此凸环结构只在引物内形成。这些结构是由引物与模板序列之间的杂交相互作用所形成的,并本领域技术人员可以通过理论计算进行设计(见j.santaluciajr.etal.(2004)“thethermodynamicsofdnastructuralmotifs”annu.rev.biophys.biomol.struct.33:415–440)。所公开的ω引物提供在各种应用中需要的特征和/或性质。本发明一方面是利用两个分开的结合部来平衡引发特异性和结合强度。在一个典型应用中,,5p臂的设计比3p臂具有更高的结合能,从而使得5p臂可以启动和维持结合而3p臂能检查聚合酶延伸序列特异性。一个成功的引发反应要求两个分开的部分杂交。这减少了偏离靶引发的几率,并产生特异性更高的引物设计。5p臂的结合强度比3p臂的结合强度可以大1.1倍,或1.2倍或1.3倍或1.4倍或1.5倍1.6倍或1.7倍,或1.8倍或1.9倍或2倍或2.5倍,或3倍或5倍或10倍。5p臂的结合强度比3p臂的结合强度可以大在1.1到100倍,1.1至50倍,1.1至25倍,1.1至20倍,1.1至15倍,1.1至10倍,1.1至5倍,1.5到100倍,1.5至50倍,1.5至25倍,1.5至20倍,1.5至15倍,1.5至10倍,1.5至5倍,2至100倍,2至50倍,2至25倍,2至20倍,2至15倍,2至10倍,2至5倍,5至100倍,5至50倍,5至25倍,5至20倍,5至15倍,5至10倍,5至5倍之间。这种设计的另一方面是使ω引物在指定引发位点具有耐受一定模板序列变异的能力。此特征是用于覆盖一般人群非常理想的检测。例如,千人基因组计划报告在人群中3800万单核苷酸多态性(snp)变体的验证单体型图(the1000genomesprojectconsortium(2012)“anintegratedmapofgeneticvariationfrom1,092humangenomes”nature491:56–65)。这意味着在60亿个二倍体核苷酸长的人类基因组中,平均不到200个核苷酸就有一个snp变体。当前版本人类ncbidbsnp数据库包含7400万snp基因型变体,意味着在三十亿个核苷酸长的人类基因组中平均每40个核苷酸中有一个snp变体。snp是人类基因组中最丰富的变体形式。这些变体对pcr引物的设计提出了挑战,因为设计用于参考靶序列的引物恰好被应用到在对应引发区包含一个或多个变体的实际靶样品中时可能不起作用。由于一般人群中变体密度高,理想的引物是能够容忍变体,从而最大化靶序列中引发可到达的区域。本公开的一方面将变体纳入引物设计的考虑范围。在设计ω引物中,设计该引物的5p臂长度应足够长,使得5p臂和所有预期变体的靶等位基因之间的结合在相应的反应温度是稳定的。该3p臂长度应足够长,以实现与参考靶等位基因的稳定结合。因此,5p臂用作锚,3p臂检查引发位点的正确性。这种设计策略显著扩大了靶序列中引发的可到达区域,产生了宽容变体的引物,也产生了特异性高的引发。5p臂可以是至少长10个核苷酸长,或至少15个核苷酸长或至少20个核苷酸长,或至少25个核苷酸,或至少30个核苷酸长,或至少35个核苷酸长或至少40个核苷酸长,或至少45个核苷酸长,或至少50个核苷酸长,或至少55个核苷酸长或至少60个核苷酸长,或至少65个或至少70个核苷酸长,或至少75个核苷酸长或至少80个核苷酸长,或至少85个核苷酸,或至少90个核苷酸长,或至少95个核苷酸长或至少100个核苷酸长,或至少125个核苷酸长,或至少150个核苷酸长,或至少200个核苷酸长或至少250个核苷酸长。5p臂可以是10至200个核苷酸之间的长度,或长度为10到150个核苷酸之间,或长度为10至100个核苷酸之间,或长度为10到90个核苷酸之间,或10至80个核苷酸之间长,或10至70个核苷酸之间的长度,或长度为10到60个核苷酸之间,或长度为10到50个核苷酸之间,或长度为10到40个核苷酸之间,或在10至30个核苷酸之间长度,或10至20个核苷酸之间的长度,或长度为15到200个核苷酸之间,或长度为15到150个核苷酸之间,或长度为15至100个核苷酸之间,或者是在15至90个核苷酸之间长度之间,或15至80个核苷酸长度,或为15至70个核苷酸之间的长度,或长度为15到60个核苷酸之间,或长度为15到50个核苷酸之间,或长度为15到40个核苷酸之间,或15至30个核苷酸之间的长度,或15至20个核苷酸之间的长度,或20至200个核苷酸之间的长度,或长度为20到150个核苷酸之间,或长度为20至100个核苷酸之间,或者20到90个核苷酸之间的长度,或长度为20到80个核苷酸之间,或在20到70个核苷酸之间的长度,或长度为20到60个核苷酸之间,或者是20至50个核苷酸长度,或长度为20到40个核苷酸,或20至30个核苷酸之间的长度,或在长度25到200个核苷酸之间,或在25至150个核苷酸之间的长度,或在长度在25至100个核苷酸之间,或者是长度在25至90个核苷酸之间,或长度为25到80个核苷酸之间,或在25至70个核苷酸之间的长度,或在长度在25至60个核苷酸之间,或在长度在25至50个核苷酸之间,或长度在25至40个核苷酸之间,或为25到30个核苷酸之间的长度,或在长度为30至200个核苷酸之间,或在30至150个核苷酸之间的长度,或在长度为30至100个核苷酸之间,或为30到90个核苷酸之间长度,或长度为30到80个核苷酸,或者30到70个核苷酸之间的长度,或在长度为30至60个核苷酸之间,或者在长度为30至50个核苷酸之间,或者在长度为30至40个核苷酸之间,或者是35至200个核苷酸之间的长度,或在长35至150个核苷酸之间,或者在长度35至100个核苷酸之间,或者在长度为35到90个核苷酸之间,或者在长度35到80个核苷酸之间,或35到70个核苷酸之间的长度,或在长度在35至60个核苷酸之间,或者在长度35至50个核苷酸之间,或者在长度为35到40个核苷酸之间,或者是在长度在40至200个核苷酸之间,或40至150个核苷酸之间的长度,或在长度在40至100个核苷酸之间,或者在长度为40到90个核苷酸之间,或者在长度为40到80个核苷酸之间,或在40到70个核苷酸之间的长度,或可在长度为40到60个核苷酸之间,或者是在长40至50个核苷酸之间。3p臂可以是至少长5个核苷酸,或至少6个核苷酸长或至少7个核苷酸长,或至少8个核苷酸,或至少9个核苷酸长,或至少10,或至少11个核苷酸长或至少12个核苷酸长,或至少13个核苷酸长,或至少14个核苷酸长,或至少15个核苷酸长或至少20个核苷酸长,或至少25个或至少30个核苷酸长,或至少35个核苷酸长或至少40个核苷酸长,或至少45个核苷酸,或至少50个核苷酸长,或至少55个核苷酸长或至少60个核苷酸长,或至少65个核苷酸长,或至少70个核苷酸长,或至少80个核苷酸长或至少90核苷酸长。3p臂可以为5至100个核苷酸之间的长度,或长度为5至90个核苷酸之间,或长度为5至80个核苷酸之间,或在5至70个核苷酸之间的长度,或在5至60个核苷酸之间长度,或5到55个核苷酸之间的长度,或长度为5至50个核苷酸之间,或在5至45个核苷酸之间的长度,或长度为5至40个核苷酸之间,或者是在5至35个核苷酸之间长度,或5至30个核苷酸之间的长度,或长度为5至25个核苷酸之间,或长度为5至20个核苷酸之间,或长度为5至15个核苷酸之间,或者是在长度为5至10个核苷酸之间,或者是6至100个核苷酸之间的长度,或长度为6到90个核苷酸之间,或长度为6到80个核苷酸之间,或者是6到70个核苷酸之间的长度,或长度为6到60个核苷酸之间,或者是在长度为6到55个核苷酸,或核苷酸长度在6到50之间,或6至45个核苷酸之间的长度,或长度为6到40个核苷酸之间,或长度为6到35个核苷酸之间,或者6至30个核苷酸之间的长度,或长度为6至25个核苷酸之间,或长度为6到20个核苷酸之间,或长度为6到15个核苷酸之间,或者是6到10个核苷酸长,或长度为7至100个核苷酸,或7至90个核苷酸之间的长度,或在长度为7至80个核苷酸之间,或7到70个核苷酸之间的长度,或在长度为7至60个核苷酸之间,或者是7到55个核苷酸长度,或7至50个核苷酸的长度,或7至45个核苷酸之间的长度,或在长度为7到40个核苷酸之间,或者在长度为7到35个核苷酸之间,或者是7到30个核苷酸之间长度,或7至25个核苷酸之间的长度,或在长度为7到20个核苷酸之间,或者在长度为7至15个核苷酸之间,或者是在长度为7至10个核苷酸之间,在10至100个核苷酸之间长度,或10至90个核苷酸之间的长度,或长度为10到80个核苷酸之间,或在10至70个核苷酸之间的长度,或长度为10到60个核苷酸之间,或在10至55个核苷酸之间长度之间,或长度为10到50个核苷酸长度,或10至45个核苷酸之间的长度,或长度为10到40个核苷酸之间,或长度为10到35个核苷酸之间,或者是长度为10至30个核苷酸之间,或10至25个核苷酸之间的长度,或长度为10到20个核苷酸之间,或长度为10到15个核苷酸之间,或介于15至100个核苷酸之间的长度,或在长度15到90个核苷酸之间,或者长度为15至80个核苷酸,或15至70个核苷酸之间的长度,或长度为15到60个核苷酸之间,或长度为15到55个核苷酸之间,或长度为15到50个核苷酸之间,或15到45个核苷酸之间的长度,或长度为15到40个核苷酸之间,或长度为15到35个核苷酸之间,或长度为15到30个核苷酸之间,或长度为15至25个核苷酸之间,或者在长度为15至20个核苷酸之间。关于公开的ω引物设计,存在各种实施例。例如,引物可以包括由两个或多个环分开的三个或更多个结合部分。理想的是将引物的结合特征分配到环部分所分开的两个或更多个分段的结合部分,以达到提高引发特异性,调节结合强度,插入或掺入特异性序列,和/或获得其他理想的功能的目的。环可以是至少5个核苷酸长,或至少6个核苷酸长,或至少7个核苷酸长,或至少8个核苷酸,或至少9个核苷酸长,或至少10,或至少11个核苷酸长,或至少12个核苷酸长,或至少13个核苷酸长,或至少14个核苷酸长,或至少15个核苷酸长,或至少20个核苷酸长,或至少25个核苷酸或至少30个核苷酸长,或至少35个核苷酸长或至少40个核苷酸长,或至少45个核苷酸,或至少50个核苷酸长,或至少55个核苷酸长,或至少60个核苷酸长,或至少65个核苷酸长,或至少70个核苷酸长。环可以是7至100个核苷酸之间的长度,或在长度为7至90个核苷酸之间,或者在长度为7至80个核苷酸,或长度为7至70个核苷酸之间,或在长度为7至60个核苷酸之间,或者是在长度为7至55个核苷酸之间,或7至50的核苷酸长度之间,或7至45个核苷酸之间的长度,或在长度为7到40个核苷酸之间,或者在长度为7到35个核苷酸之间,或者7至30个核苷酸之间的长度,或在长度为7至25个核苷酸之间,或者在长度为7到20个核苷酸之间,或者在长度为7至15个核苷酸之间,或者是在长度为7至10个核苷酸之间,介于长度为10至100个核苷酸之间,或10至90个核苷酸之间的长度,或长度为10到80个核苷酸之间,或在10至70个核苷酸之间的长度,或长度为10到60个核苷酸之间,或者是10到55个核苷酸之间长度,或长度为10到50个核苷酸之间,或在10至45个核苷酸之间的长度,或长度为10到40个核苷酸之间,或长度为10到35个核苷酸之间,或长度为10到30个核苷酸,或10至25个核苷酸之间的长度,或长度为10到20个核苷酸之间,或长度为10到15个核苷酸,或长度为12到100个核苷酸之间,或介于12至90个核苷酸之间长度,或长度为12到80个核苷酸之间,或12至70个核苷酸之间的长度,或长度为12到60个核苷酸之间,或长度为12到55个核苷酸之间,或长度为12到50个核苷酸之间,或12到45个核苷酸之间的长度,或长度为12到40个核苷酸之间,或长度为12到35个核苷酸之间,或长度为12到30个核苷酸之间,或者是在长度为12到25个核苷酸之间,或12至20个核苷酸之间的长度,或在长度12至15个核苷酸之间,介于15至100个核苷酸之间的长度,或长度为15到90个核苷酸之间,或15至80个核苷酸之间的长度,或长度为15到70个核苷酸之间,或15至60个核苷酸之间的长度,或长度为15到55个核苷酸之间,或长度为15到50个核苷酸之间,或在15到45个核苷酸之间的长度,或介于长度为15至40个核苷酸之间,或15至35个核苷酸之间的长度,或长度为15到30个核苷酸之间,或长度为15至25个核苷酸之间,或者是长度为15至20个核苷酸之间。本发明的示例性实施例是一种高特异性杂交探针。设计探针(ω探针)序列,这样当与靶序列杂交时,形成侧翼有ω臂的一个或多个ω环。图2显示了示例性的ω探针220,包括间隔区201,ω臂1到3,202、204、206、203、以及ω环1,203和环2,205。根据特定的应用,间隔区201是可选的,并且可以连接到探针的3’或5’的末端。对于需要探针附着于表面的应用,通常优选具有通过间隔区的附着。在一些实施例中,间隔区201包括一个或多个核苷酸。在一些实施例中,该间隔区201包括一个或多个非核苷酸部分。在一些实施例中,非核苷酸部分包括但不限于至少一个c3烷基间隔区,至少一个乙二醇间隔区和至少一个1’,2’-双脱氧核糖。在一些实施例中,该ω臂202,204或206包括至少一个核苷酸。在一些实施例中,核苷酸的数目在1和100之间。在一些实施例中,核苷酸的数目在3和60之间。在一些实施例中,核苷酸的数目在5和40之间。在一些实施例中,每个环203或205包括一个或多个核苷酸。在一些实施例中,核苷酸的数目在1和100之间。在一些实施例中,核苷酸的数目在3和60之间。在一些实施例中,核苷酸的数目在5和40之间。在一些实施例中,每个环203或205包括一个或多个非核苷酸部分。在一些实施例中,非核苷酸部分包括但不限于至少一个c3烷基间隔区,至少一个乙二醇间隔区,和至少一个1’,2’-双脱氧核糖。在一些实施例中,每一个非核苷酸环203或205的长度在1和200个分子键之间。在一些实施例中,每个环203或205的长度在5和100个分子键之间。在一些实施例中,每个环203或205的长度在5和60个分子键之间。从热力学角度,探针210和相应的靶模板序列220之间的结合反应中,ω臂202,204和206与模板220的结合,降低自由能,并稳定结合,而ω环203和205拉紧,增加自由能,结合不稳定。在一个实施例中,设计臂的长度和环,这样很好地平衡由于臂引起的自由能降低和由于环引起的自由能增加,以当探针与目的靶序列在预定的杂交条件下杂交时,产生稳定的ω形结构。当靶包含一个或多个变体核苷酸时,探针-靶结合的原始结构就不再不稳定,使得结合减少。所公开的ω探针的设计,与包含一个连续的靶互补结合区段的常规杂交探针设计相比,一个显著的优点是不牺牲特异性,延长探针长度的能力。一般情况下,核酸结合测定法报告了与相应的探针杂交的靶的数目。这数目通常是通过测量含有探针的表面或含有探针的溶液上结合密度得到的。平衡的结合密度是由结合自由能确定。自由能越低,平衡结合密度越高(miuraelal.(2005)“anovelstrategytodesignhighlyspecificpcrprimersbasedonthestabilityanduniquenessof3’-endsubsequences”bioinformatics214363-4370)。常规探针的结合自由能随探针长度的增加而增加。当常规探针是长的时(例如超过35个核苷酸),靶中短的变体如snp只会在探针-靶结合中产生一小部分自由能。因此,常规探针的序列特异性随着探针长度的增加而减小。通过比较,对ω探针-靶结合自由能由ω环调节,即使整体结合的长度长,也可以通过设计维持在一定的理想水平。因此,ω探针保留了对更大范围探针-靶结合长度的高特异性。整个ω结构的自由能在杂交条件下要低于零的。在本说明书的“计算方法”部分描述了ω引物的自由能计算。ω探针的一个示例性实施例是微阵列测定法,该测定法可以涉及一个或多个检测方法,包括但不限于用荧光染料标记靶序列做常规荧光检测,或用供体和受体荧光基团在环核苷酸上或附近标记ω探针进行fret(荧光共振能量转移)检测。这些额外的标记和检测方法是由buzdin在a.buzdin和s.lukyanov(2007)“stem-loopoligonucleotidesashybridizationprobesandtheirpracticaluseinmolecularbiologyandbiomedicine”nucleicacidshybridization,chapter14,85–96,springer)中描述了。接力pcr本发明另一方面是关于靶扩增的方法,被称为接力pcr。一个完整的pcr过程包括由两个功能不同,但依次连接的反应阶段,即靶筛选和文库扩增,即图3中所示的阶段1和阶段2反应。本发明的方法的显著优点是,利用程序化pcr仪在单个管中进行两个功能不同的反应阶段,没有任何其他人工操作。用于接力pcr的反应混合物包括2组引物、模板以及适合的聚合酶、核苷酸和pcr缓冲液。第一组引物包括至少一对特异性引物301和302。一般来说,每对引物勾勒出目的靶区域304。示例性特异性引物对包括特异性引物1,301和特异性引物2。302。一示例性的特异性引物340(一个“常规”的引物)包括3’末端特异性区段341(10~80个核苷酸之间,优选长度为12至50个之间的核苷酸)和5’末端通用区段342(10和80个核苷酸之间,优选长度为12至50个之间的核苷酸)。每个特异性引物的特异性区段的序列与相应的靶序列或模板的相应部分基本上相同或互补。第二组引物包括至少一对通用引物。在一个示范性实施例中,使用了一对通用引物,包括通用引物1,321、通用引物2,322。示例性的通用引物350包含3’末端的通用区段351和5’末端尾区段352。特异性引物1,301和通用引物1,321的通用区段的序列基本相同。特异性引物2,302和通用引物2,322的通用区段的序列基本相同。在优选实施例中,通用区段的序列基本不与样本dna序列的任何部分杂交。在优选的实施例中,通用引物的浓度显著高于相应的特异性引物的浓度。在某些实施例中,通用引物和对应特异性引物的摩尔浓度比为至少50、100、500、1,000、5,000、10,000、50,000或更大。各个引物的序列设计、引物合适的浓度、通用引物和特异性引物浓度比以及聚合酶的选择将作为明确的反应条件和应用要求进行说明。图3示意性概述了接力pcr的示范性实施例。在该图中,阶段1是靶筛选阶段,有两个热循环,并涉及一对特异性引物:特异性引物1,301和特异性引物2,302。第一轮循环中,特异性引物1,301和特异性引物2,302在相应的模板300上延伸,产生互补链的两个复制的第一轮循环产物序列311和312。这里,模板是其上发生聚合酶延伸反应和产生互补复制的核酸序列。在这个说明书中,术语“模板”,“靶序列”和“样品dna”是根据描述的上下文,可互换使用。通过延伸反应,特异性引物的通用区段掺入到第一轮循环产物序列中。第二轮循环中,特异性引物1,301和特异性引物2,302在相应的第一轮循环产物序列311和312上延伸,产生互补链的两个复制的第二轮循环产物序列323和324。第二轮循环产物序列323和324侧翼有在5'末端的特异性引物1和2的通用区段325和326,在3'末端的特异性引物的互补通用区段327、328。第二轮循环产物序列是余下循环中扩增的两端侧翼的靶序列。一般地,通用引物被设计为不与样品dna序列的任何部分杂交,也不在第一轮和第二轮循环反应中涉及。图3中,第三轮循环到第n轮循环构成第二阶段,其中靶序列的文库被扩增。从第三轮循环开始,两端侧翼的靶序列323和324变得可用,它们携带互补通用区段327和328与该通用引物321和322杂交,并进行扩增反应。当多对特异性引物和一对通用引物都参与其中,第二轮循环结束后,所有相应的靶序列侧翼具有相同对的通用区段,并在第三轮循环到第n轮循环(最后一轮循环)之间由一对通用引物进行扩增。通用引物与特异性引物的高浓度比确保通用引物在第2阶段的扩增反应中占主导地位。所公开的接力pcr方法的一方面消除了进行多个人工pcr循环和产物纯化的需要。这是以前从靶特异性引物扩增转换成通用引物扩增使用的(例如z.linetal.1996,“multiplexgenotypedeterminationatalargenumberofgeneloci”proc.natl.acad.sci.93:2582-2587;k.e.varleyetal.2008,“nestedpatchpcrenableshighlymultiplexedmutationdiscoveryincandidategenes”genomeres.18:1844–1850和j.leamonetal.2012,“methodsandcompositionsformultiplexpcr”美国专利申请公布us2012/0295819a1)。所公开的接力pcr方法的另一方面将靶特异性引物从参与扩增过程中排除(图3中第轮循环到第n轮循环),从而最小化特异性引物相关的扩增偏差。所公开的接力pcr反应的一个特点就是第一阶段限制特异性引物直接从原始样品模板产生侧翼靶序列的功能,以最小化第二阶段中特异性引物堆靶序列扩增反应的贡献。当有2对或更多对特异性引物参与时,这最小化了引物依赖性的扩增的产率变化。反应条件和引物设计对所公开的接力pcr反应的结果有重要影响。对于多重pcr的应用,本发明的不同反应条件是与常规pcr反应和已知的多重pcr反应的变化中所使用的靶特异性引物浓度相比,使用了显著更低的靶特异性引物浓度。可以在比以前所用的特异性引物浓度更低的特异性引物浓度下进行接力pcr反应。常规pcr翻译中,引物浓度为100nm和5000nm之间(见dieffenbachetal.,pcrprimer:alaboratorymanual,coldspringharborlaboratorypress,2003)。在已知的多重pcr反应的变化中,靶特异性引物浓度为10nm和400nm之间(见henegariuetal.1997,“multiplexpcr:criticalparametersandstep-by-stepprotocol”biotechniques23:504-511;j.brownieetal.1997,“theeliminationofprimer-dimeraccumulationinpcr”nucleicacidsres.25:3235-3241;k.e.varleyetal.2008,“nestedpatchpcrenableshighlymultiplexedmutationdiscoveryincandidategenes”genomeres.18:1844–1850和b.freyetal.(2013),“methodsandamplificationoftargetnucleicacidsusingamulti-primerapproach”美国专利申请公布us2013/00045894a1)。通过比较,在本发明的示例性实施例中,如实施例i到iii所示,每一特异性引物的浓度为1nm或更低,且每个通用引物的浓度为500nm。本发明的另一个不同的反应条件是第1轮和第2轮热循环中使用显著延长的退火时间与低浓度特异性引物结合。在常规pcr和已知多重pcr反应的变化中,退火时间在10s到2min之间。通过比较,在本发明的示例性实施例中,如实施例i到iv所示,在第1轮和第2轮热循环中,使用30min和4h之间的延伸时间。在浓度为1nm或更低的特异性引物浓度,使用先前报道的退火时间30s,得到的pcr产物量低,低于实验中常规的琼脂糖凝胶电泳测量的检测限(例如b.freyetal.2013,“methodsandamplificationoftargetnucleicacidsusingamulti-primerapproach”美国专利申请公开us2013/00045894a1)。本发明的另一个不同的反应条件是,在第1轮和第2轮循环中,使用高的退火温度和长的延伸时间相结合。这些示范性的反应条件只代表本发明的某些方面,而不应该被解释为以任何方式限制本发明。具体应用的精确的反应条件,由本领域技术人员根据本说明书整体和所包括的参考文献的教导确定。本发明的一个方面是ω引物的使用,有利地做为特异性引物在接力pcr中的使用。图4a示意了使用ω引物401和402做为特异性引物的接力pcr的示例性概括实施例。在每个引物中,3p臂403和5p臂405都是具有靶序列特异的。换句话说,臂序列是与对应的模板序列400的引发部分互补的。ω引物的环部分404和441用作特异性引物的通用区段。通用引物421、422和445包括在3’末端的通用区段446和在5’末端的尾段447。相应的通用引物440和ω引物445的通用区段441和446的序列基本相同。如图4a所示,一个完整的接力pcr过程包括两个阶段,阶段1的靶筛选和阶段2的扩增。靶筛选阶段包括2个热循环和涉及一对特异性引物:特异性引物1,401和特异性引物2,402。在第一轮循环中,特异性引物1,401和特异性引物2,402在相应的模板400上延伸,产生互补链的两个复制的第一轮循环产物序列410。原始特异性引物402包括第一轮循环产物410的5’部分。特异性引物402的原始3p臂403,环404,和3p臂405分别成为第一轮循环产物410的特异性引发区段411,通用引发区段412和5p臂的尾段413。在第二轮循环中,特异性引物1,401和特异性引物2,402在相应第一轮循环产物序列410上延伸,产生互补链的两个复制的第二轮循环产物序列420。第二轮循环产物序列420在靠近5’端的部分中侧翼有通用区段412和414以及在靠近3’端的部分中侧翼有互补的通用区段423和424。互补的通用区段423和424与通用引物421和422互补,并用作第3轮循环至第n轮循环的pcr扩增反应的引发位点。在某些实施例中,涉及多对特异引物,所有引物对共享相同对的环序列,从而使靶序列文库在一对通用引物下进行扩增。第一轮循环产物和第二轮循环产物的5p臂的尾部413绕过通用引物422和423的文库扩增。结果,最终扩增产物430只保留原始的ω特异性引物的3p臂部分。尽管图4a中的ω引物具有内环,其他环形式的ω引物也可应用到接力pcr中。作为另一个示例性实施例,图4b描绘接力pcr过程中使用凸环ω引物做为特异性引物451和452。在图4a和图4b所示的靶筛选和文库扩增过程的原理基本相同。结合ω引物和接力pcr的示范性应用是测序测定法的靶富集。该组合为应用带来了显著和独特的好处。事实上,扩增产物中只保留原始ω特异引物的3p臂部分403和453是非常可取的,因为它提供了最小化扩增的靶序列430和480中特异性引物部分431和481长度的机会。这是通过使用长5p臂和短3p臂,同时保持ω引物和相应模板在对应的退火温度下足够稳定结合来实现的。为了测序使用,从特异性引发部分431或481之间的天然部分432和482的测序读取来自于测试样品的天然序列,,而从特异性引物部分431和481的测序读取由使用的引物确定。由于测序过程的读取长度是有限的,缩短特异性引物部分431和481是非常可取的,以便最大化天然序列的有用的读取长度。在一些实施例中,通用引物1的尾部区段352,447和497还包括至少一个条形码部分。在一些实施例中,通用引物2的尾部区段352,447和497还包括至少一个条形码部分。在一些实施例中,通用引物1和通用引物2两者的尾部区段352,447和497还包括至少一个条形码部分。。条形码部分的设计和使用参考bystrykhlv(2012)generalizeddnabarcodedesignbasedonhammingcodes.plosone7(5):e36852.doi:10.1371/journal.pone.0036852。在一些实施例中,至少一个ω引物包含两个或多个环。在图5中所示的一个示范性实施例中,特异性引物501包括接力引发环502和插入环503。接力引发环502作为特异性引物的通用区段。在第一轮反应中特异性引物1,501延伸并形成第一轮循环产物511。这导致插入环503作为插入片段513掺入到产物中。剩余的循环继续以同样的方式进行上述有关图4a和图4b中的接力pcr过程。在过程完成时,插入片段533掺入到最终产物530中。这种区段引物的示范性应用包括但不限于诱变,基因敲除,基因敲进,特征标记,蛋白质工程和基因治疗。利用接力pcr的靶富集的一些实施例包括靶筛选,该靶筛选使用通用引物pcr的单一引物延伸和扩增:单一引物延伸接力pcr。图6描述了一个示例性应用的实施例,其包括样品制备和接力pcr过程。样品制备涉及片段化和衔接头连接。这一过程公知为分子生物学领域,是由n.arneson等人((2008)“whole-genomeamplificationbyadaptor-ligationpcrofrandomlyshearedgenomicdna(prsg)”coldspringharbprotoc)和d.bentley等人((2008)“accuratewholehumangenomesequencingusingreversibleterminatorchemistry”nature45653-59)以及相关的配套文件进行了详细的阐述。一个示例的样品制备从双链dna模板600的随机片段化为短区段开始。可以从不同来源,包括不限于基因组dna和rna来源的双链cdna产生双链dna序列。双链dna片段化可以使用一个或多个各种众所周知的过程包括雾化、超声处理和酶促消化。雾化可以使用商业产品雾化器和产品说明书(来自于lifetechnologies(grandisland,ny))。超声处理可以使用各种商业产品之一来完成,例如聚焦超声仪(来自于covarisinstrument(woburn,ma))。酶促消化法可使用商业试剂盒dsdnafragmentase(来自于neb(ipswich,ma))。修复dna片段末端使用ultratmendrepair/da-tailingmodule(来自于neb(ipswich,ma))。利用t4dna连接酶(来自于neb(ipswich,ma))连接,添加衔接头603到片段到末端补齐片段。就得到衔接头侧翼的片段604。衔接头603是双链dna序列,包括正链寡核苷酸601和负链寡核苷酸602。在一些实施例中,正链寡核苷酸601和负链寡聚核苷酸602是完全彼此互补的。在一些实施例中,正链寡核苷酸601和负链寡聚核苷酸602彼此部分互补。在一些实施例中,正链寡核苷酸601比负链寡核苷酸602短,且与负链寡核苷酸602的3’部分基本互补。在一些实施例中,正链寡核苷酸601的3’末端的修饰核苷酸阻断聚合物延伸反应的寡核苷酸。示例的修饰核苷酸包括但不限于双脱氧胞苷,倒置的dt,3’氨基修饰基因和3’生物素。在一些实施例中,正链寡核苷酸601的5’末端被磷酸化。在一些实施例中,负链寡核苷酸602的3’末端是da突出端。图6的下部分描述了单一引物延伸接力pcr过程,其中每个靶序列由单一特异性引物的延伸反应筛选,然后通过一对通用引物pcr扩增。在示例性实施例中,反应混合物包括衔接头侧翼片段604作为样本模板,一个或多个ω引物为靶特异性引物610,通用引物1,611,通用引物2,612和聚合酶,所有混合在pcr缓冲液中。每个ω引物包括3p臂615,至少一个环616和5p臂617。3p臂615和5p臂617的序列是根据预定的靶序列从起始dna模板600设计的。环616包含与通用引物2,612的3’部分具有大致相同序列的的部分。通用引物1,611的3’部分的序列1和负链寡核苷酸602的选定部分基本上相同。在一些实施例中,负链寡核苷酸602的选定部分覆盖了负链寡核苷酸602的大部分。在一些实施例中,负链寡核苷酸602的选定部分覆盖了负链寡核苷酸602减去与正链寡核苷酸601重叠部分的其大部分。在第一轮循环反应中,特异性引物610结合到相应的靶序列片段614,然后其被延伸以产生第一轮循环产物620。在第二轮循环中,通用引物1,611结合第一轮循环产物620的衔接头621部分,然后延伸以产生第二轮循环产物630。第二轮循环产物630包含通用的引发部分631,与特异性引物610的环616互补,并结合通用的引物2,612,以方便在剩下循环中的pcr扩增。从第三轮循环到第n轮循环,筛选的靶序列在配对的通用引物1,611和通用引物2,612下被扩增,产生产物640。尽管图6的示例性实施例描述了ω引物做为特异性引物,可替代的实施例中,可以在单一引物延伸接力pcr中使用常规引物作为特异性引物。在接力pcr中使用常规引物,已在上述图3被描述。常规引物物不是ω引物;在一些实施例中,常规引物有结合区段,但没有环和(或)多余的错配(即它与其靶序列一一对应)。分离在一些实施例中,接力pcr产物溶液进一步纯化,以将pcr产物430、330、480、530和640从其余的反应混合物中分离出来。各种建立的pcr反应纯化方法和商业试剂盒可用于此目的。这些包括但不限于标准化磁珠,来自于axygenbiosciences(unioncity,ca),pcr纯化柱(来自于qiagen(valencia,ca)),和切胶纯化。图7说明了本发明的纯化方法的示例性实施例。起始混合物包括靶序列700、通用引物1(701)、通用引物2、片段707和聚合酶,全部在pcr缓冲液中。靶序列700侧翼有与通用引物1和通用引物2相同或互补的引发区段,并设计为由引物扩增。片段707不携带引发区段,不期望由引物扩增。这起始混合物是类似于图6第一轮循环反应结束时的反应混合物。通用引物2(702),包括引发区段,修饰基因区段704和标记区段705。修饰基因区段704的功能是阻止聚合酶延伸反应通过区段和/或方便在位置上酶促、化学或光裂解。修饰基因区段704的示例性实施例包括但不限于至少一个c3烷基间隔区,至少一个乙二醇间隔区,至少一个光裂解的间隔区,至少一个1’,2’-双脱氧核糖和至少一个脱氧尿苷。这些修饰基因掺入到寡核苷酸中在核酸合成领域众所周知,这可以由商业供应商如集成dna技术公司(coralville,ia))进行。在一个实施例中,通用引物2(702)包括六乙二醇间隔区作为修饰基因区段704,其阻止聚合酶延伸反应通过区段和进入标记区段705中。结果pcr产物710含有单链突出端标记区段715。在一些实施例中,标记区段705包含一个寡核苷酸。在一些实施例中,标记区段705包括至少一个结合部分。示例性的结合部分是生物素。在一些实施例中,标记区段705包括寡核苷酸和结合部分的组合。在一些实施例中,结合部分是连接到标记区段705寡核苷酸的5’末端。在一些实施例中,将一种或多种上述纯化试剂盒或方法应用于该pcr产物溶液,以除去聚合酶和残留的单链引物。在一些应用中,如单一引物延伸接力pcr,,片段717有类似pcr产物710的平均大小,和需要额外的或不同的纯化过程中以去除片段717。图7所示的下部分说明所公开的纯化方法的示例性实施例涉及直接捕获pcr产物720。在pcr产物溶液中加入探针722接枝的珠721,或是引物去除的pcr产物溶液。探针722的序列基本上互补标记区段715的序列,并有利于通过杂交,由珠721捕获pcr产物720。可选地,溶液盐浓度可以调节,以确保足够的杂交。盐包括但不限于氯化钠、柠檬酸钠、磷酸钠、氯化钾、磷酸钾、氯化铵、tris氯化物,和/或两种或多种盐的混合物。在室温下或在预定的温度下,在溶液中搅拌和孵育,提高捕获产率。用杂交缓冲液洗涤珠721去除片段717,残留聚合酶和游离引物,同时保留杂交的聚合酶链反应产物720。为完成纯化,将珠721放在洗脱缓冲液中,并将缓冲液温度升高,以从珠721释放pcr产物720。寡核苷酸固定以形成探针722接枝珠721和使用珠捕获核酸序列在生物学领域是众所周知的。一个示范性实施例中使用myonetmstreptavidinc1(来自于lifetechnologies(grandisland,ny))作为珠721。探针722和标记区段705的互补对的序列设计分享了杂交探针设计的许多原理和程序,这是本领域技术人员所熟悉的。该序列以这样的方式设计,这样在结合过程期间和在杂交缓冲液中获得该对之间的稳定杂交,但是在释放过程期间获得该对之间的变性。一般情况下,结合和/或杂交缓冲液具有较高的盐浓度,至少为50mm,100mm,500mm,1m,2m,或更高。洗脱缓冲液具有相对低的盐浓度,至多100mm,50mm,10mm,5mm,1mm或更低。一般情况下,结合温度低于洗脱温度。温度是根据相应缓冲溶液中的探针722/标记705对的tm(熔化温度)决定的。优选的结合温度在结合或杂交缓冲液中探针/标记对的tm以下。优选的洗脱温度在洗脱缓冲液中的探针/标记对的tm以上。在一些实施例中,在洗脱过程后,pcr产物730保持双链。在一些实施例中,结合温度至多40℃、35℃、30℃、25℃、22℃或更低。洗脱温度至少为30℃,35℃,40℃,45℃,50℃,55℃,或更高。在序列设计中的其他注意事项包括最小化探针722序列与任何靶序列的交叉杂交,并最小化涉及标记区段705的二级结构。所公开方法的替代实施例涉及pcr产物720的间接捕捉。在一些实施例中,探针722不是预先接枝到珠721,而是包含配体部分,如生物素。珠721涂覆有捕获部分,如链霉亲和素。在实施例中,第一,游离探针722与标记区段715杂交。然后,捕获部分覆盖的玻珠721被添加到杂交溶液中以捕捉pcr产物720。其余的过程类似于上面所描述的。上述纯化方法的一个方面是具有单链突出端的pcr产物的直接形成。突出端可以用来促进纯化。该方法使得在温和的变性条件下释放双链产物730。相比之下,常规的生物素化引物,没有修饰基因区段704和标记区段705,会产生平末端。虽然pcr产物可以被链霉亲和素包被的玻珠捕获,但是产物的释放需要强有力的变性条件(见lifetechnologies的myonetmstreptavidinc1用户手册,grandisland,ny),在该条件下,pcr产物的双链结构也会变性。本公开描述了在用于基于表面簇的测序中文库制备和簇形成的新改进方法中包含pcr产物的单链突出端的使用(d.bentleyetal.(2008)“accuratewholehumangenomesequencingusingreversibleterminatorchemistry”nature45653-59)。图8示意性地表示出了新的方法。图8的顶部描述了pcr的靶扩增,这类似于图7,并已在上述描述了。靶扩增过程如图8所示,是一个通用pcr过程,包括下代测序中序列文库的制备(mardiser(2008)"next-generationdnasequencingmethods",annurevgenomicshumgenet9:387–402)。所公开的方法的一个特点是产生含有单链标记815的pcr产物810。在一些实施例中,通用引物2(802)包括引发区段803、修饰基因区段804和标记区段805,并在图3至图6的接力pcr过程中作为通用引物1,321、421、471、521和611或作为通用引物2,322、422、472、523和612使用。所公开的方法还包括含有pcr产物810的单链标记815固定到基底,用于进行化学和/或生化反应。作为一个示范性的例证,在图8中,基底826与探针822,表面引物1(823),和表面引物2(824)接枝。根据应用,基底826可以用玻璃、硅、聚合物、金属或任何其他合适的材料制成。接枝部分,探针822,表面引物1(823)和表面引物2(824)是预定序列的寡核苷酸。在一些实施例中,接枝部分还包括间隔区827,以将寡核苷酸连接到基底表面。接枝寡核苷酸对到基底表面是公知技术,已在文献中描述(b.joosetal.(1997)“covalentattachmentofhybridizableoligonucleotidestoglasssupports”analbiochem,24796-101;y.rogersetal.(1999)“immobilizationofoligonucleotidesontoaglasssupportviadisulfidebonds:amethodforpreparationofdnamicroarrays”analbiochem,26623-30;d.bentleyetal.(2008)“accuratewholehumangenomesequencingusingreversibleterminatorchemistry”nature45653-59,supplementaryinformation)。pcr产物810的固定开始于单链标记815,引导821和探针822的共杂交。引导821是一段寡核苷酸序列,一部分序列与标记815互补,一部分序列与探针822互补。在一些实施例中,进行共杂交是首先以足够高的引导821浓度,将引导821溶液应用于基底826上,以在基底上基本上与所有探针822杂交,再洗去溶液中任何多余的引导821。这一步在基底表面上产生引导/探针对。以足够高的pcr产物810浓度(阈浓度),通过应用pcr产物以基本上与所有的引导/探针对杂交来完成共杂交。在一些实施例中,一旦杂交,标记815的5’端堆栈到探针的3’端,并且在两端之间没有缝隙。在一些实施例中,标记815的5’末端被磷酸化,而探针822的3’末端为羟基。标记815和探针822可以通过连接共价地连接在一起。在一些实施例中,连接是通过使用neb的t4连接酶(ipswich,ma)完成。标记815,探针822和引导821的序列设计主要考虑因素包括熔化温度,序列唯一性和分子内折叠。标记/引导和探针/引导对在相应缓冲液中的熔化温度应该大大高于杂交和连接温度,从而可以达到和/或保持在两个反应条件下足够高的杂交产率。在一些实施例中,探针/引导对的熔化温度高于标记/引导对的熔化温度。所有这三个序列在反应条件下应具有最小折叠。所有这三个序列应该足够不同于表面引物1(823)和表面的引物2(824)的序列,因此,不会发生与两个表面引物的交叉杂交。此外,杂交温度,连接温度和缓冲组合物应以这样的方式设计,使得pcr产物810的双链结构在这两个反应中仍然保持稳定。更准确的序列和反应条件的设计将在本公开的稍后部分中描述。替代方法可用于连接标记815和探针822。在一些实施例中,标记815的5’末端是叠氮化物基团,探针822的3’末端是炔基团。标记815和探针822然后使用由r.kumar等人描述的点击化学连接到一起((2007)“template-directedoligonucleotidestrandligation,covalentintramoleculardnacircularizationandcatenationusingclickchemistry”j.am.chem.soc.129,6859-6864)。在一些实施例中,标记815和探针822共价结合后,系统包括基底和固定的pcr产物820,被放入变性缓冲液中,洗涤去除双链828和引导821。在一些实施例中,该变性缓冲液包含高浓度的甲酰胺。其结果是单链产物830共价地连接到基底826。在一些实施例中,产物830,表面引物1(823)和表面引物2(824)进一步发生酶促和/或化学反应,以产物830作为起始模板延伸表面引物,从而在起始产物830附近产生产物830的额外拷贝,这形成产物830的表面簇。然后,产物830的平行拷贝作为模板进行测序。表面簇的形成和测序过程在d.bentley等人发表的文章以及相关补充资料中有详细描述((2008)“accuratewholehumangenomesequencingusingreversibleterminatorchemistry”nature45653-59),这些文献由此以它们整体并入为参考文献。在一些实施例中,表面引物1(823)的序列与侧翼1(831)的序列基本上是互补的。在一些实施例中,表面引物2(824)的序列基本上与侧翼2(833)的序列相同。在某些实施例中,表面引物1(823)的表面密度基本上与表面引物2(824)的表面密度相同。表面密度的定义是每单位表面积的固定的分子数。在一些实施例中,表面引物1(823)和表面引物2(824)的表面密度显著高于探针822的表面密度。在某些实施例中,表面引物1(823)(或表面引物2(824))与探针822的表面密度比率是至少1000、10000、100000、1000000、10000000、100000000或更大。在某些实施例中,表面密度比率由在基底表面的接枝制备中所用的探针822、引物1(823)和引物2(824)混合液的相对浓度所控制。所公开的方法的某些方面包括向基底表面添加探针和表面引物以及使用探针捕获的序列作为簇形成的种子。在本领域实践中(d.bentleyetal.(2008)“accuratewholehumangenomesequencingusingreversibleterminatorchemistry”nature45653-59),基底表面只能接枝有以基本上相同表面密度存在的两种引物。表面簇的种子是由一个表面引物和一个pcr产物的侧翼经引物的聚合酶延伸杂交形成的。当表面引物被密集地群集在基底表面上时,种子密度是由pcr产物的溶液浓度控制。在正常条件下,终簇密度与种子密度成比例。在一个平行的测序过程中,从每个簇发出的信号被用来推导出种子模板的序列。为了从单个簇获得可靠的信号检测,需要足够的相邻簇之间的距离,否则,从相邻的簇发射的信号是不可分离的,将产生错误的序列读取。另一方面,为了最大化读取通量的目的,提高簇密度是可取的。存在最佳的簇密度。在目前的实践中,通过仔细控制簇溶液中的pcr产物浓度,实现了光学簇密度。这个过程是耗时和受测量仪器以及人类操作失误的影响。通过比较,在本发明的方法中,通过基底制造商在基底表面制备期间固定的探针密度或表面引物与探针的比率来控制簇密度。其结果是,对于用户点的簇制备,只要用户使pcr产物浓度超过阈值,所产生的簇密度会永远停留在固定值附近。在一些实施例中,使用不同序列的多个探针822。当多个样品平行测序时,这种布置特别有用。每个样品进行靶扩增时,都使用的单一的一对通用引物1(801)和通用引物2(802)。不同样本的通用引物2(802)包括不同序列的不同标记805。每个单一标记805通过单一引导821与单一探针822配对。多个标记,探针和引导的设计考虑必须包括序列之间的交叉杂交最小化。在一些实施例中,不同探针的数目至少为1、2、4、8、12、16、32、48、64、96或更多。在一些实施例中,所有探针中每个不同探针的表面密度基本上是相同的。在一些实施例中,不同探针的表面密度是不同的。在某些实施例中,选定组探针的表面密度设置在预定值,而使得其余的探针的表面密度基本上相等。在某些实施例中,表面引物1(823)(或表面引物2(824))与联合探针822的表面密度比率是至少1000、10000、100000、1000000、10000000、100000000或更大。在一些实施例中,通用引物1(801)包括至少一个条形码部分。在一些实施例中,通用引物2(802)的引发段包括至少一个条形码部分。引物中条形码组合是用来识别序列读取中样本来源。在一个示例性实施例中,在多样品平行测序的簇制备中使用包含多探针的基底,应用涉及基底826的所有引导821的溶液混合物以形成引导/探针对。洗去多余的引导821。然后,所有样本中含有pcr产物810的标记815的溶液混合物被施加到基底上,以完成标记/引导/探针共杂交。连接,变性和簇形成如上所述进行。其结果是,每个样品簇的数目与基底表面上可用的样本特异性探针数量是直接相关的,只要样品pcr产物的浓度高于阈值,就与样品pcr产物的浓度几乎无关。通过比较,目前多个样本的平行测序中现有技术的簇制备方法需要所有所涉及样品的精确样品数量标准化和精确控制的总pcr产物浓度。本方法的自限性的特点在易操作、结果的一致性和整体过程的牢靠性方面比目前的方法具有显著的优势。聚合酶的选择在本发明的一些实施例中,扩增产物通过使用一种或多种dna聚合酶,经聚合酶链反应形成。在一些实施例中,该聚合酶可以是热稳定聚合酶。在一些实施例中,该聚合酶可以是热启动聚合酶。在一些实施例中,该聚合酶可以是一种高保真性聚合酶。在一些实施例中,该聚合酶可以是重组聚合酶。在一些实施例中,聚合酶可以是商业化的产品如taqdna聚合酶(lifetechnologies,grandisland,ny),accuprimetmtaqdna聚合酶(lifetechnologies,grandisland,ny),amplitaqdna聚合酶(lifetechnologies,grandisland,ny),taqdna聚合酶(newenglandbiolabs,ipswich,ma),dna聚合酶(newenglandbiolabs,ipswich,ma),deepventtmdna聚合酶(newenglandbiolabs,ipswich,ma),hotstartflexdna聚合酶(newenglandbiolabs,ipswich,ma),high-fidelitydna聚合酶(newenglandbiolabs,ipswich,ma),pfuturbocxhotstartdna聚合酶(agilent,santaclara,ca),pfuultraiifusionhsdna聚合酶(agilent,santaclara,ca),kapahifipcrkits(kapabiosystems,wilmington,ma)。在本发明的一些实施例中,使用一个或多个热启动聚合酶用于最小化潜在的偏离靶的扩增和引物二聚体的形成。在一些实施例中,有利地使用一种新的热稳定的高保真聚合酶,其缺乏链置换活性和5’至3’核酸酶活性。缺乏链置换和5’至3’的核酸酶活性对多重扩增的应用有利,在多重扩增中两个或多个靶区域是串联或位置接近。通过使用这些聚合酶,引物与串联区域中间的结合不会被移位或退化。这类新的热稳定的高保真聚合酶的商业产品包括,但不限于hotstartflexdna聚合酶(newenglandbiolabs,ipswich,ma)和high-fidelitydna聚合酶(newenglandbiolabs,ipswich,ma)。反应条件反应条件一般由所涉及的聚合酶、引物的热动力学性质,靶序列的特性和多重pcr多重性决定。一个接力pcr过程包括2个功能不同的反应阶段,这可以需要不同的反应条件。具体应用的具体反应条件可以由本领域技术人员通过遵循下面的具体考虑,在本说明书后面的实施例以及该专利说明书的教导和参考文献而确定。接力pcr的第一阶段的功能是在特异性靶序列侧翼产生通用区段。应该设计相应的反应条件以达到下列目的:产生最高可能靶向复制产率,最小化偏离靶向的模板复制,以及最小化引物二聚体的形成。如图3至图5所示,第一阶段反应包括第1和2轮循环反应。在图6中,第一阶段包括第1轮循环反应。每个循环包含三个热步骤,包括变性,退火,和延伸。第1轮循环和第2轮循环的退火条件是需要考虑的关键因素。以下原则是用来指导的注意事项。随着特异性引物的数量增加,引物二聚体的形成概率增大((见u.landegrenetal.(1997)“lockedontarget:strategiesforfuturegenediagnostics”ann.med.,29:585–590)。另一方面,基于化学反应的热动力学和动力学(见i.tinocoetal.(1995)“physicalchemistry:principlesandapplicationsinbiologicalsciences”prenticehallcollegediv;3rdedition),随着引物浓度下降,平衡产物浓度以及引物-引物的速率以及靶向引物-模板杂交的相互作用下降。此外,基于核酸杂交的热动力学原理(见j.santaluciajr.etal.(2004)“thethermodynamicsofdnastructuralmotifs”annu.rev.biophys.biomol.struct.33:415–440),杂交稳定性随杂交温度的降低和互补序列长度的增加而增加。总的来说,随着扩增反应复杂性增加和/或特异性引物数量增加,每个单独引物浓度应降低,以减小引物-二聚体形成的可能性;同时,引物浓度不应大幅下降,从而所需的引物和模板之间的相互作用不会减弱。在这样的情况下,引物的浓度降低,退火时间应增加,以弥补引物和模板之间减少的相互作用率。一般优选高的退火温度用于获得高的引物特异性和最小化不期望的引物-引物杂交。当选定的聚合酶具有高3’至5’的核酸酶活性时,优选高的退火温度以避免在长时间的退火周期中引物的净降解。通常期望发现接近峰值聚合酶活性温度的最佳的退火温度,在10℃范围内。从相应的聚合酶供应商获得峰值聚合酶活性温度。在本说明书的实施例i到实施例iv提供了本发明的示例性的反应条件。对于高复杂度的扩增应用,最佳反应条件比之前已知的条件变化明显。靶特异性引物浓度为0.001nm或更低,0.01nm或更低,0.1nm或更低,1nm或更低,2nm或更低,3nm或更低,4nm或更低,或5nm或更低。在本发明的一些实施例中,靶特异性引物浓度是约从0.0001nm到10ηm,或约从0.0001nm到5nm,或约从0.0001nm到4nm,或约从0.0001nm到3nm,或约从0.0001nm到2nm,或约从0.0001nm到1nm,或约从0.0001nm到0.1nm,或约从0.0001nm到0.01nm,或约从0.0001nm到0.001nm,或约从0.001nm到10ηm,或约从0.001nm到5nm,或约从0.001nm到4nm,或约从0.001nm到3nm,或约从0.001nm到2nm,或约从0.001nm到1nm,或约从0.001nm到0.1nm,或约从0.001nm到0.01nm,或约从0.01nm到10ηm,或约从0.01nm到5nm,或约从0.01nm到4nm,或约从0.01nm到3nm,或约从0.01nm到2nm,或约从0.01nm到1nm,或约从0.01nm到0.1nm,或约从0.1nm到10ηm,或约从0.1nm到5nm,或约从0.1nm到4nm,或约从0.1nm到3nm,或约从0.1nm到2nm,或约从0.1nm到1nm,或约从1nm到10ηm,或约从1nm到5nm,或约从1nm到4nm,或约从1nm到3nm,或约从1nm到2nm。退火时间可以大于5分钟,大于10分钟,大于20分钟,大于30分钟,大于40分钟,大于50分钟,大于60分钟,大于70分钟,大于80分钟,大于90分钟,大于100分钟,大于110分钟,大于120分钟,大于130分钟,大于140分钟,大于150分钟,大于160分钟,大于170分钟,大于180分钟,大于190分钟,大于200分钟,大于210分钟,大于220分钟,大于230分钟,大于240分钟。退火时间可以是约5分钟至500分钟,或约5分钟至400分钟,或约5分钟至300分钟,或约5分钟至250分钟,或约5分钟至200分钟,或约5分钟至150分钟,或约5分钟至100分钟,或约5分钟至50分钟;或约10分钟至500分钟,或约10分钟至400分钟,或约10分钟至300分钟,或约10分钟至250分钟,或约10分钟至200分钟,或约10分钟至150分钟,或约10分钟至100分钟,或约10分钟至50分钟;或约20分钟至500,或约20分钟至400分钟,或约20分钟至300分钟,或约20分钟至250分钟,或约20分钟至200分钟,或约20分钟至150分钟,或约20分钟至100分钟,或约20分钟至50分钟;或约30分钟至500,或约30分钟至400分钟,或约30分钟至300分钟,或约30分钟至250分钟,或约30分钟至200分钟,或约30分钟至150分钟,,或约30分钟至100分钟,或约30分钟至50分钟;或约40分钟至500,或约40分钟至400分钟,或约40分钟至300分钟,或约40分钟至250分钟,或约40分钟至200分钟,或约40分钟至150分钟,,或约40分钟至100分钟,或约40分钟至50分钟;或约50分钟至500,或约50分钟至400分钟,或约50分钟至300分钟,或约50分钟至250分钟,或约50分钟至200分钟,或约50分钟至150分钟,或约50分钟至100分钟;或约60分钟至500,或约60分钟至400分钟,或约60分钟至300分钟,或约60分钟至250分钟,或约60分钟至200分钟,或约60分钟至150分钟,,或约60分钟至100分钟。优先选择高的退火温度。退火温度可大于50℃,或大于60℃,或大于65℃,或大于70℃,或大于75℃,或大于80℃,或大于85℃,或大于90℃。退火温度可以约从50℃至95℃,或约从50℃至90℃,或约从50℃至85℃,或约从50℃至80℃,从约从50℃至75℃,或约从50℃至70℃,或约50℃至65℃,或约从50℃至60℃;或约从55℃至95℃,或约从55℃至90℃,或约从55℃至85℃,或约从55℃至80℃,或约从55℃至75℃,或约从55℃至70℃,或约从55℃至65℃,或约从55℃至60℃;或约从60℃至95℃,或约从60℃至90℃,或约从60℃至85℃,或约从60℃至80℃,或约从60℃至75℃,或约从60℃至70℃,或约从60℃至65℃;或约从65℃至95℃,或约从65℃至90℃,或约从65℃至85℃,或约从65℃至80℃,或约从65℃至75℃,或约从65℃至70℃;或约从70℃至95℃,或约从70℃至90℃,或约从70℃至85℃,或约从70℃至80℃,或约从70℃至75℃;或约从75℃至95℃,或约从75℃至90℃,或约从75℃至85℃,或约从75℃至80℃;或约从80℃至95℃,或约从80℃至90℃,或约从80℃至85℃;或约从85℃至95℃,或约从85℃至90℃,或约从90℃至95℃。通过比较,在先前已知的条件下,靶特异性引物的浓度为10nm至400nm,退火时间为10秒和2分钟之间和建议低退火温度(见henegariuetal.(1997)“multiplexpcr:criticalparametersandstep-by-stepprotocol”biotechniques23:504-511;j.brownieetal.(1997),“theeliminationofprimer-dimeraccumulationinpcr”nucleicacidsres.25:3235-3241;以及k.e.varleyetal.(2008)“nestedpatchpcrenableshighlymultiplexedmutationdiscoveryincandidategenes”genomeres.18:1844–1850;b.freyetal.(2013)“methodsandamplificationoftargetnucleicacidsusingamulti-primerapproach”美国专利申请公开us2013/00045894a1)。在一些实施例中,所有特异性引物的浓度基本相同。在一些实施例中,不同特异性引物的浓度是不同的。在一些实施例中,在预定水平制备所选数目引物的浓度,并靶向样品序列的预定部分用于控制目的和/或用于任何其它期望目的。在一些实施例中,通用引物的浓度显著高于相应的特异性引物的浓度。在一些实施例中,通用引物的浓度至少为50nm,100nm,200nm,500nm,1000nm,2000nm,5000nm,或更高。通用引物的浓度可约从10nm至5000nm,或约从10nm至1000ηm,或约从10nm至500nm,或约从10nm至250nm,或约从10nm至100nm;或约从25nm至5000nm,或约从25nm至1000ηm,或约从25nm至500nm,或约从25nm至250nm,或约从25nm至125nm,或约从25nm至100ηm,或约从25nm至50nm;或约从50nm至5000nm,或约从50nm至1000ηm,或约从50nm至500nm,或约从50nm至250nm,或约从50nm至100ηm。在一些实施例中,通用引物与所对应的特异性引物的的摩尔浓度比率可以至少为10、20、50、100、200、500、1000、2000、5000、10000、20000、50000、500000或更大。通用引物与所对应的特异性引物的的摩尔浓度比率可以在10至5000000之间,或从10至500000,或从10至50000,或从10至5000,或从10至500,或从10至50;或从50至5000000,或从50至500000,或从50至50000,或从50至5000,或从50至500;或从500至5000000,或从500至500000,或从500至50000,或从500至5000。在一些实施例中,特异性引物的浓度至多50nm,20nm,5nm,2nm,1nm,0.5nm,0.2nm,0.1mm,0.05nm,0.02nm,0.01nm,0.001nm,或更低。在一些实施例中,第1轮循环和第2轮循环的退火温度的选择应用用来平衡聚合酶3'至5’核酸酶活性与聚合酶活性。在一些实施例中,第1轮循环1和第2轮循环的退火温度这样选择使得所用的聚合酶的核酸酶活性和聚合酶活性的组合结果在退火阶段不会引起特异性引物的大幅降解。在一些使用ω引物和含有3’至5’外切酶活性的聚合酶的实施例中,第一阶段的退火步骤包括两个阶段,阶段1和阶段2。阶段1温度接近或为聚合酶延伸温度,而阶段2温度低于阶段1。阶段1退火时间或持续时间比阶段2长。每个ω引物的5p臂设计得足够长,在阶段1温度,5p臂序列与相应的模板序列稳定杂交。而3p臂设计得足够短,使得在阶段1的温度,3p臂大部分保持自由形式未结合)。这样设计该条件,以最小化在阶段1长退火时间期间ω引物的3'核酸外切酶消化,设计阶段1长退火时间以使引物与模板序列高比率杂交。3p臂的长度被设计成这样一种方式,使得在第2阶段的退火温度下3p臂与模板杂交。对于第1轮循环和第2轮循环延伸步骤和阶段2热循环的反应条件可以用通用pcr条件设定,就像在对应聚合酶供应商的产品说明书中发现的一样。在一些实施例中,当涉及多个高gc含量的靶序列时,延伸时间可以延长。在一些实施例中,延伸时间是至少15秒,30秒,60秒,90秒,120秒或更多。在本说明书的实施例中提供了示例性的反应条件。在一些实施例中,在进行多重pcr的样品中加入加标(spike-in)对照。在一个示例性实施例中,加标对照是已知序列的化学合成核酸。在另一个示例性实施例中,通过提取已知序列的生物核酸来获得加标对照。在另一个实施例中,加标对照是化学合成核酸以及提取已知序列的生物核酸的混合物。加标对照的序列可从选自双链核酸序列、单链核酸序列,含有与通用引物3’端匹配的3’和5’端以及在通用引物下进行扩增的序列,以及这样序列,其含有不与通用引物3’端匹配,而是与相应的特异性引物匹配的3’和5’端,,并可通过特异引物进行复制。在一些实施例中,加标对照包括多个不同gc含量的核酸序列。在一个实施例中,gc含量变化从15%到85%,或从15%到80%,或从15%到75%,或从15%到70%,或从15%到65%,或从15%到60%;或从20%到85%,或从20%到80%,或从20%到75%,或从20%到70%,或从20%到65%,或从20%到60%;或25%到85%,或从15%到80%,或从25%到75%,或从25%到70%,或从25%到65%,或从25%到60%。具体应用中,加标对照的gc含量的实际范围由具体应用的本领域技术人员所决定。在一些实施例中,对含有加标对照的样品扩增产物进行定量分析用于优化反应条件。在一些实施例中,对含有加标对照的样品扩增产物进行定量分析用于质量控制。在一些实施例中,对含有加标对照的样品扩增产物进行定量分析用于进行定量标准化。引物制备在一些实施例中,引物可以通过常规化学合成制成((l.j.mcbrideetal.(1983)“aninvestigationofseveraldeoxynucleosidephosphoramiditesusefulforsynthesizingdeoxyoligonucleotides”tetrahedronletters,24:245248)。在一些其他的实施例中,引物是由化学合成在微阵列上制成((x.zhouetal.(2004)“microfluidicpicoarraysynthesisofoligodeoxynucleotidesandsimultaneouslyassemblingofmultiplednasequences”nucleicacidsres.32:5409-5417;x.gaoetal.“methodandapparatusforchemicalandbiochemicalreactionsusingphoto-generatedreagents”.us专利us6,426,184;gao,x.,zhang,h.,yu,p.,leproust,e.,pellois,j.p.xiang,q.,zhou,x..“linkersandco-couplingagentsforoptimizationofoligonucleotidesynthesisandpurificationonsolidsupports”.us7,211,654,au2002305061;gao,x.,zhou,x.,cai,s.-y,you,q.,zhang,x.“arrayoligomersynthesisanduse”wo2004/039953))。微阵列的合成具有每一个序列成本低的优点,其中在需要至少有10,50,100,500,5000,10000,50000,或100000引物序列的高多重pcr的应用中特别有利。产品名称为oligomixtm的微阵列合成的寡核苷酸混合物可以从lcsciences(houston,tx)商购获得。在一些实施例中,在被用作pcr引物之前,扩增合成的寡核苷酸。当化学合成的规模较低,如微阵列上化学合成的情况,首选扩增。图9所示为通过扩增产生特异性引物的示例性实施例。引物前体模板900使用上述微阵列的常规方法化学合成。每个引物前体模板900包括具有多个功能区段的寡核苷酸,包括5p侧翼901,5p臂902,环903,3p臂904和3p侧翼905。在3p侧翼区段905的5’端为da(脱氧腺苷)核苷酸。为了示例目的,图9所示的引物前体模板900是专为制备ω引物设计的。然而,通过简单地用常规引物区段(例如图3中的特异性区段341和通用区段342)替换5p侧翼901和3p侧翼905之间部分,模板设计也可以用于制备常规引物,。2对引物的制备,制备引物1(906)和制备引物2(907)用于pcr扩增。制备引物1(906)序列与5p侧翼901序列基本相同。制备引物2(907)序列与3p侧翼905的序列基本上是互补的。此外,制备引物2(907)的3’端是du(脱氧尿苷)核苷酸。虽然只有一个引物的前体模板900是绘制在图9中,扩增的目的是同时扩增多个引物的前体模板900,以产生多个靶筛选和文库扩增的特异性引物。所有单个引物前体模板900都有相同对的5p侧翼和3p侧翼区段,但在中间有特异性引物部分。含有du引物的使用需要pcr聚合酶有能力读完尿嘧啶。可接受的聚合酶,包括但不限于neb的热启动taqdna聚合酶(ipswich,ma)和agilent的pfuturbocx热启动dna聚合酶(santaclara,ca)。在混合模板的pcr扩增过程中,应采取措施最小化序列偏差,以及避免pcr产物交叉杂交。热启动聚合酶是首选。制备引物1(906)和制备引物2(907)的熔化温度优选比聚合酶制造商所建议的最高延伸温度高5℃或以上,从而使pcr退火和延伸温度可以最大化。首选较低的pcr循环数。在一些实施例中,循环数最多为25、24、23、22、21、20、19、18、17、16、15或更低。完成前体扩增之时,双链前体910产生。下一步是去掉前体的帽子,其中前体910负链919的du在udg/eda(尿嘧啶dna糖基化酶/乙二胺)溶液中被消化。另外,du可使用neb的usertm(尿嘧啶特异性切除试剂)酶消化(ipswich,ma)。在某些实施例中,在消化后应用纯化处理以除去消化酶和/或改变缓冲组合物。纯化方法包括但不限于axygenbiosciences的标准化珠(unioncity,ca),qiagen的pcr纯化柱((valencia,ca),),并切胶纯化。在消化的前体920中,负链929的3p臂924的5’端没有帽子结构。图9描述了片段927与前体920的正链928杂交,在某些实施例中,在消化过程中或在纯化过程中,片段927是从链928解离的。图9的下部说明了如何激活被消化的前体转化为特异性引物,以及如何将激活过程与靶筛选和文库扩增结合在一起。在三个功能不同的过程中,引物激活,靶筛选和扩增可在单一的管中进行。示例性的起始反应混合物包括消化的前体930,制备引物1(936),模板941,通用引物1(942),引物2(943),一个或多个dna聚合酶、和含有dntp的pcr缓冲液。消化的前体930和消化的前体920相同,制备引物1(936)与引物1(906)相同。消化的前体930和制备引物1(936)的适宜浓度的类似于特异性引物和通用引物的浓度,这已在“反应条件”部分被描述。在如上所述的一个类似的方式中,其他组份被用来进行接力pcr,其合适的浓度已在“反应条件”部分被描述。反应是在pcr仪上进行的。在第一轮循环中,制备引物1(936)与负链939退火,然后延伸形成活化的特异性引物940,带有可延伸的3p臂。如图3,4,5和6所示,从第二轮循环开始,以上面描述的相同方式进行作为接力pcr过程进行的反应过程。此方法的一个方面是活性特异性引物的原位生产。该方法的优点是过程简单和所涉及的步骤最少。图10所示的是通过扩增产生特异性引物的另一个示例性实施例。pcr模板1000包括特异性引物的spc引物区段1002,5p侧翼区段1001和3p侧翼1003。3p侧翼1003的5’端为da(脱氧腺苷)核苷酸。spc引物区段1002可以为常规特异性引物序列,ω特异引物,或任何核苷酸序列。2个制备引物,制备引物1(1004)和制备引物2(1005)用于pcr扩增。制备引物1(1004)包括3’部分,其序列与5p侧翼1001的序列基本相同。制备引物2(1005)包括3’部分,其序列与3p侧翼1003的序列基本互补。制备引物2(1005)还包括3’末端的du(脱氧尿苷)。在一些实施例中,最好是有一个或多个du纳入到制备引物2(1005)的中部分。在一些实施例中,多重pcr模板1000包括在过程中,该过程每个模板包含不同的spc引物区段1002,但是相同的5p侧翼区段1001和相同的3p侧翼区段1003。适用的条件已经在上述图9的过程中描述。完成pcr扩增时,双链产物1010产生。下一步是去除双链pcr产物1010的负链1019的du。在一些实施例中,在udg(尿嘧啶dna糖基化酶)溶液中du被移除或被消化,以除去尿嘧啶,然后在eda(乙二胺)溶液中剪去核苷酸骨架。在一些实施例中,du使用usertm(尿嘧啶特异性切除试剂)酶消化(neb(ipswich,ma))。这两种消化过程产生具有优选的磷酸化5’末端的负链1029。在一些实施例中,消化后应用纯化处理,以除去消化酶和/或改变缓冲组合物。纯化方法包括但不限于标准化珠(axygenbiosciences(unioncity,ca)),pcr纯化柱(qiagen(valencia,ca)),并切胶纯化。在一些优选的实施例中,一个或多个du已掺入制备引物2(1005)中部分中,制备引物2剩余片段太短而不能保持在正链1028上,得到单链3p侧翼1023。下一步是通过去除单链3p侧翼1023末端补齐产物1020。末端补齐过程的要求是为了产生平端双链产物1030,其具有与spc引物1002的3’端相同的正链1038的3’端,以及为了保持负链1039的磷酸化5’末端完整。在一个实施例中,末端补齐是利用neb的t4dna聚合酶进行(ipswich,ma)。t4dna聚合酶具有3’-5’外切酶活性,没有5’-3’核酸外切酶的功能。末端补齐的产物1030中正链1038具有羟基3’末端,并且可在接力pcr中作为活性特异性引物。通过去除活性引物的反义链,可以提高特异性引物的效率。最后一步是去除负链1039。在一些实施例中,通过酶促移除负链1039。在一个实施例中,用λ核酸外切酶(neb(ipswich,ma))消化负链1039。本制备过程的最终产物为单链特异性引物1040。图11说明了通过扩增产生特异性引物的另一个实施例。pcr模板1100包括特异性引物的spc引物区段1102,5p侧翼区段1101和3p侧翼1103。在一些实施例中,3p侧翼区段1103包括一个或多个由一个或多个限制酶识别的限制酶切位点。spc引物区段1102可以为常规的特异性引物序列,ω特异性引物,或任何核苷酸序列。两个制备引物,制备引物1(1104)和制备引物2(1105)用于pcr扩增。制备引物1(1104)包括3’部分,其序列与5p侧翼1101的序列基本相同。制备引物2(1105)包括3’部分,其序列与3p侧翼1103的序列基本互补。制备引物2(1105)还包含一个或多个限制酶切位点。在一些实施例中,多重pcr模板1100包括在过程中,该过程每个模板包含不同的spc引物区段1102,但是相同的5p侧翼区段1101和相同的3p侧翼区段1103。与图9和图10中的pcr反应相比,图11中的pcr的成分不需要du,因此该方法中使用的pcr聚合酶不需要具有读完尿嘧啶的能力。合适的聚合酶已在上述“聚合酶的选择”部分中描述了。在混合模板的pcr扩增过程中,应采取措施最小化序列偏差,避免pcr产物交叉杂交。首选热启动聚合酶。。制备引物1(1104)以及制备引物2(1105),优选以这样一种方式设计,从而使pcr退火和延伸温度可在最高可能的水平使用。本公开的后续部分将提供引物设计的详细说明。首选较低的pcr循环数。在一些实施例中,循环数最多为25、24、23、22、21、20、19、18、17、16、15或更低。完成前体扩增之时,双链产物1110产生。下一步是限制酶切消化。这一步是将双链pcr产物1110的正链1118上的3p侧翼1103移除,以暴露spc引物区段的3’端。在一些实施例中,使用一个或多个限制酶来进行消化。在一些实施例中,使用iis型限制酶(a.pingoudetal.(2001)“structureandfunctionoftypeiirestrictionendonucleases”nucleicacidsres.293705-3727)。在一些实施例中,从商业供应商获得限制酶,如neb(ipswich,ma)。示例性的限制酶,包括但不限于bspqi,sapi,bsai-hf,bpuei,bfuai,acui和btsi(neb(ipswich,ma))。在一些实施例中,在设计模板1100、制备引物1及制备引物2中考虑的因素之一是限制3p侧翼1103和制备引物2中识别位点序列,避免在模板1100和制备引物1(1104)中任何其他部分中的识别位点序列。在优选实施例中,在消化产物1120中,正链1128的3’端与spc引物1102的3’端相同。在优选的实施例中,在消化产物1120中,负链1129的5’端磷酸化,正链1128的3’端为羟基基团。消化产物1120中的正链1128具有羟基3’末端,可作为活性的特异性引物在接力pcr中使用。通过去除活性引物的反义链,可以提高特异性引物的效率。最后一步是去除负链1129。在一些实施例中,酶促地移除负链1129。在一个实施例中,用λ核酸外切酶(neb(ipswich,ma))用来消化负链1129。本制备过程的最终产物为单链特异性引物1130。引物与靶序列的设计在一些实施例中,特异性引物的形式包括两个功能不同的部分,如图3所示。3’区是靶特异性片段和5’区是通用片段。在一些实施例中,该特异性引物的ω形式包括三个功能不同的部分,如图4a和4b所示。3p臂是第一靶特异性区段;环部分是通用区段;5p臂是第二靶特异性区段。在一些实施例中,如图3、4a、4b、5所示,通用引物包括两部分。3’区是通用区段,5’区是尾区段。在一些实施例中,使用一对通用引物,通常由通用引物1和通用引物2组成。其中,通用引物1和2分别由通用区段1和2与尾区段1和2组成。在使用常规形式的特异性引物的实施例中,每对特异性引物都包含第一特异性引物和第二特异性引物,第一特异性引物由通用区段1和第一靶特异性区段组成,第二特异性引物由通用区段2和第二靶特异性区段组成。在使用ω形式的特异性引物的实施例中,每对特异性引物应该包含第一特异性引物和第二特异性引物,第一特异性引物由通用区段1、第一3p臂和第一5p臂组成;第二特异性引物由通用区段2、第二3p臂和第二5p臂组成。在某些实施例中,可以使用2个或更多对通用引物。然后,相应组的特异性引物对与具有相应组特异性引物的每对通用引物一起使用。每组通用引物和特异性引物的组分和相互关系与上述段中描述的相同。一般情况下,特异性引物和通用引物的通用区段序列的设计表现出以下特点:(i)它们应该与目的样品中任何预期或可疑的没有实质的杂交;(ii)它们应该与自身或彼此之间没有实质的杂交;以及(iii)它们应该没有稳定的二级结构。此外,通用引物的3’端应产生基本稳定的杂交。这通常是通过在3’端具有足够的gc含量实现的。通用引物的通用区段的gc含量和长度应该按照熔化温度不低于第二阶段的热循环退火温度进行设计。在一些实施例中,本发明用于制备大规模平行测序应用的样品。设计通用引物的尾区段以容纳克隆乳液扩增,克隆桥式扩增,和/或序列模板制备过程中所涉及的任何其它反应(e.r.mardis(2008)“next-generationdnasequencingmethods”annu.rev.genomicshum.genet.9:387–402)。在一些实施例中,通用引物的尾区段包含dna条形码。一般情况下,常规形式特异性引物的靶特异性区段的序列与目的靶序列的选定部分基本上是相同或互补的。所选择的序列表现出以下特点:(i)它应该与目的样本中预期或可疑序列的任何其它部分具有最小化杂交;(ii)它应该与其它特异性引物,与通用引物和与自身没有实质的杂交;(iii)引物的3’末端应与模板产生基本稳定的杂交;(iv)它应该是足够长的,以与相应的模板在相应的退火条件下形成基本稳定的杂交;以及(v)特异性引物整体(包括特异性区段和通用区段)应该没有稳定的二级结构。选择ω特异性引物的3p臂和5p臂的序列的原则类似于上述段落中描述的常规形式特异性引物。下列是直接针对ω形式的形成的额外考虑。(i)环应该与目的样品中预期或可疑序列的任何部分具有最小的杂交;(ii)3p臂5p臂长度应足够长以使得3p臂5p臂与对应模板在相应的退火条件下同时基本稳定杂交。这些结构是由引物与模板序列之间相互杂交作用形成,本领域技术人员可以通过理论计算设计它们(见j.santaluciajr.etal.(2004)“thethermodynamicsofdnastructuralmotifs”annu.rev.biophys.biomol.struct.33:415–440)。在一些实施例中,特异性引物和/或通用引物中含有修饰的核苷酸,用于性能改进和/或特定的应用。在一个实施例中,引物的3’端由硫代磷酸修饰的核苷酸组成。一方面,硫代磷酸修饰的核苷酸抑制核酸外切酶降解。在一些实施例中,3’端的硫代磷酸修饰的核苷酸的数量至少是1,2,3或更多。计算方法本发明描述了严格的pcr引物设计方法。该方法的一个方面是使用严格热动力学计算以定量预测引物性能,包括靶向引物/靶结合系数,引发效率,偏离靶延伸概率,和引物二聚体的形成概率。所公开的方法在定量方面,相比于使用按经验制定的分数来决定引物选择的现有技术的引物设计方法和/或工具具有明显的改进(a.untergasseretal.(2012)“primer3-newcapabilitiesandinterfaces”nucleicacidsres.40:e115)。该方法的另一个方面是变体耐受引物的设计,以实现对来自一般群体的样品的加强的性能。图12描绘了所公开的计算过程的流程图。在以下的说明中,大多采用人类基因组相关序列作为例子。然而,所公开的方法可以用于设计任何物种,包括人工靶序列的引物。该说明书的这一部分涉及生物信息学和热动力学。所公开的方法可供本领域技术人员来设计pcr引物。引物设计方法的步骤1是制备引物结合模板序列。该输入数据是用户定义的靶区域,包括数据库的版本,染色体数目,靶区域的起始和终止位置。输入数据还可以包括用户提供的靶区域中的序列变异。在优选实施例中,用户提供了单个样本或患者的序列变异。在一些实施例中,单个样本的序列变异不可用,用户可以提供组合的序列变异。在一些实施例中,用户可以通过直接提供个别的靶序列选择替代靶序列输入格式。根据靶标信息的输入数据,设计特异性引物的引物结合模板序列是从参考序列数据库中提取。每个引物结合模板序列的区域通过延伸靶区域的两端超过了相应的靶区域,以便有足够的空间可放置用于捕获整个靶区域的引物。在一些实施例中,延伸长度至少是50,75,100,125,150,200,250或更多。在一些实施例中,特异性引物用于高通量测序应用中文库的制备,选择延伸长度近似于测序运行的读长。参考序列数据库可以从各种公共和私人来源中选择。示例性的参考数据库包括人类基因组的基因组参考组合grch37和grch38,和针对小鼠基因组的grcm38,以及针对斑马鱼基因组的crcz10。在一个优选的实施例中,使用含有重复序列小写形式(soft-masked)的参考序列数据库。小写形式(soft-masking)转换重复序列为小写字母,同时保持序列的大写字母的其余部分。所提取的参考序列见图13中的参考等位基因1300。在一些实施例中,当引物设计是针对大的群体,优选在引物结合模板区域中包括变体序列,无论何时,变体数据是可用的。可以从公共或私人来源获得变异数据库。变体数据库的优选类型包括单个样本的变异。一种更为优选的变体数据库包括个体样本的单倍体型变体。对此,千人基因组计划产生和发布了人类基因组的单倍体型变体数据库(g.mcveanetal.(2012)“anintegratedmapofgeneticvariationfrom1,092humangenomes”nature,491,56-69)。另外,当单个样本的变体数据库不可用时,可以使用组合样本的变体数据库。对此,dbsnp数据库从人类snp数据库ucsc基因组浏览器中建立了138可用的组合的变体数据库。引物结合模板区域的变体序列是从现有的最佳可用变体数据库中提取。在示范性实施例中,目的区域中的变体序列是从人类千人基因组数据库中使用vcf工具提取。提取的数据包括多于1000人的单倍体型的数据。在一些实施例中,所提取的数据被编译成等位基因频率高于预定阈值的独特的单倍体型的列表。在一些实施例中,预定阈值至多10%,5%,1%,0.5%或更低。在一些实施例中,编译的单倍体型数据格式化成变体等位基因1310,1320,1330,1340,1350和1360,与引物结合模板的参考等位基因1300一致,如图13所示。这些变体等位基因在设计特异性引物序列是有用的,满足了在一个给定的群体下,一定百分比成功的需求。在另一个示例性实施例中,通过直接读取fasta文件从dbsnp数据库提取目的区域中变体序列。所提取的dbsnp序列1370与参考等位基因1300一致。在一些实施例中,当用户提供的变体序列存在,变体序列如上所述以类似的方式编译。其次,描述参考引物结合模板序列。也就是去识别任何序列特征,这在单个扩增子的起始与终止位置的确定期间将需要特殊考虑。在一些实施例中,该描述包括屏蔽(masked)重复序列和同源部分的识别。单个参考引物结合模板序列内的屏蔽重复序列的位置被提取,并在一个数据表中编译,以便后续使用。跨越单个参考引物结合模板序列以及单个参考引物结合模板序列内的同源性的部分由比对确定。在一些实施例中,使用比对工具如nih的blast进行比对。在一些实施例中,所识别的同源部分的长度至少为50、75、100、150、200、250、300、400、500或更多。在一些实施例中,所识别的同源部分的长度至少是捕获区域的平均长度。在一些实施例中,捕获区域的平均长度是基于特定应用的要求通过引物设计者预先确定。单个参考引物结合模板序列内的同源部分的位置在一个靶数据表中编译,以便后续使用。步骤2的引物设计方法是设计引物序列,并计算引物对参考和变体靶等位基因的结合特性。引物序列在预先确定组的参数指导下,经严格的热动力学计算得到。在一些实施例中,每个特异性引物的结合部分的序列由相应的参考等位基因1300的序列确定。在一些实施例中,一些特异性引物的结合部分的序列是由一些目的变体等位基因的序列(如图13中的1310、1320、1330、1340、1350或1360)所确定。本发明的方法包括一些原理计算,可以单独或组合使用来设计常规引物,ω引物,任何其他形式的引物和/或杂交探针。在一个示范性实施例中,设计了常规特异性引物。如上述描述图3的部分,常规特异性引物包括特异性区段341和通用区段342。通用区段是这个示例性实施例中的一个给定序列。下面是描述如何设计特异性区段的序列,以及如何评价其引发性能:1)固定引物的3’位置在参考引物结合模板的预定位置;2)确定模板链;3)确定特异性区段的合适长度,它在预定的模板浓度、引物浓度,缓冲盐(如na+和mg++)浓度,以及退火温度条件下,会产生与引物杂交的足够的一部分模板;4)从预定的最小长度开始,提取给定链模板的互补序列,给定的起始位置与给定的长度(在一些实施例中,最小长度至多为20个核苷酸,15个核苷酸,10个核苷酸或更少);5)将提取序列添加到通用区段的3’端以形成起始实验序列;6)计算实验序列与模板序列之间的结合自由能δg。在一些实施例中,采用近邻法计算结合自由能(见j.santaluciajr.etal.(2004)“thethermodynamicsofdnastructuralmotifs”annu.rev.biophys.biomol.struct.33:415–440)。在一些实施例中,利用计算软件包如unafold计算结合自由能((n.markhamandm.zuker(2008)unafold:softwarefornucleicacidfoldingandhybridization.inkeith,j.m.,editor,bioinformatics,volumeii.structure,functionandapplications,number453inmethodsinmolecularbiology,chapter1,pages3–31.humanapress,totowa,nj.isbn978-1-60327-428-9)。从自由能δg,模板的缔合分数fa(方程式7)是来自杂交平衡方程(方程式1和5),质量平衡方程(方程式2,3,4)与热动力学平衡常数方程(方程式6)。在该方程式中,ct,cp,和cc分别表示平衡状态下模板、引物、以及模板-引物复合物的浓度;和分别是起始模板和引物浓度;ka是平衡常数;t是退火温度;r为理想气体常数。方程式7是来源于假设方程式5和6中的起始模板浓度明显小于起始引物浓度得到的。在一些实施例中,人们可以通过求解方程式5的二阶方程来获得fa的精确解。从方程式7得到fa值可与预定的阈值模板的缔合分数fa,thr相比。在一些实施例中fa,thr至少是0.90,0.91,0.92,0.93,0.94,0.95,0.97,0.98,0.99或更多。在一些实施例中,fa,thr是约从0.90到0.99,0.91到0.99,0.92到0.99,0.93到0.99,0.94到0.99,0.95到0.99,0.97到0.99,0.98到0.99。如果fa小于fa,thr,特异性区段长度可以增加1或按预定的数量递增,重复上述计算直到fa高于fa,thr。由此产生的序列是候选引物,将进行进一步的评估。本公开的方法,相对于现有技术确定引物长度的方法和工具做了显著的改进。在所公开的方法中,在实际的退火或反应温度和反应组合物下,在计算被引物杂交的模板的分数的基础上确定引物的长度。引物的引发效率与计算的量成比例。相比之下,引物设计方法和工具的情况决定了基于引物tm的引物长度,tm是核酸的质量属性,与实际反应温度无关。在热动力学计算((j.santaluciajr.etal.(2004)“thethermodynamicsofdnastructuralmotifs”annu.rev.biophys.biomol.struct.33:415–440)中,tm涉及焓和熵。tm与自由能不是单一的关系,这涉及到焓,熵和温度,tm不能给出模板缔合分数fa的预测。按照引物设计方法的经验法则,使用引物tm以下5℃或以上3℃的退火温度,并不保证足够的或可预测的引物-模板结合。然后,进行计算以评估候选引物的引发性能。在一些实施例中,该评估包括对变体等位基因的引物结合,引物3“端结合稳定性,以及折叠对引物结合的影响。来自候选引物的参考等位基因和变体等位基因之间的杂交形成含有一个或多个非watson-crick的基序或错配的双链体,这通常导致结合自由能的增加,以及由此模板缔合分数降低(见方程式7)。在一些实施例中,当存在单个变体等位基因,候选引物和所有单个可用的变体等位基因之间的结合自由能δg采用上述的方法和工具进行计算,这包括含有错配的核酸双链体的热力学计算。然后用方程式7计算相应的模板缔合分数fa的值。挑选最低fa值作为最坏情况下候选引物的模板缔合分数fa,min。在一些实施例中,最坏的情况下,只有一个组合变体序列(例如dbsnp数据库中的一个)是可用的,使用与计算单个变体等位基因同样的方法,根据组合变体序列计算模板缔合分数fa,min。本发明公开的方法包括酶对核酸结合的影响有关的热动力学计算的考虑。按照使用供应商提供的聚合酶进行pcr的惯例,引物退火温度通常按制造商建议的水平设置。对大多数聚合酶,建议将退火温度设置为比引物tm低5℃。然而,对于新一类聚合酶,包括phusion聚合酶和q5聚合酶(都由neb,ipswich,ma提供),建议将退火温度设置为比引物tm高3℃以上。这种公开的方法需要考虑影响聚合酶和相关蛋白对结合的热动力学计算。在一些实施例中,聚合酶和相关蛋白对结合的影响被计算为一种等效盐。在示例性实施例中,根据,在涉及phusion聚合酶的反应混合物中的自由能的热动力学计算中额外添加75mm盐浓度。在一些实施例中,通过在矩阵条件下测得的实验pcr产物产率的曲线拟合得到了聚合酶和相蛋白的等效盐浓度。条件变量包括引物特异性区段长度和退火温度。在某些实施例中,实验性pcr是接力pcr。在一些实施例中,使用uv熔解曲线法测定聚合酶对结合的效果(j.santalucia(1998)“aunifiedviewofpolymer,dumbbell,andoligonucleotidednanearest-neighborthermodynamics”proc.natl.acad.sci.,95,1460-1465)。在一些实施例中,通过候选引物和参考以及变体等位基因模板之间的结合自由能的热动力学计算,进行了引物3’端结合稳定性的计算。在一些实施例中,引物3’端结合模板的引物-模板结合自由能使用常规最近邻热动力学计算。命名邻近引物的3′端或结合相应模板的3’端的自由能δgclose。然后,对于从引物的3′端开始的预定数量的碱基,或者直到达到最小自由能,从叠加能量顺序地去除最近邻自由能来计算自由能(见(see页码418-419ofj.santaluciajr.etal.(2004)“thethermodynamicsofdnastructuralmotifs”annu.rev.biophys.biomol.struct.33:415–440)。在一些实施例中,预定数量的碱基是至少1,2,3或更多。因在碱基i处开放,故命名为自由能δgopen,i。方程式8显示引物3’碱基i处打开和引物3’′封闭的结合状态之间的平衡反应。δδgi是两结合态之间的自由能差。只有当它的3'端结合到相应的模板时,该引物在延伸反应中才是有活性的。如方程式12所示,有引物3’封闭的引物-模板复合物的分数,这是来自于结合平衡方程式(方程式8和9),质量平衡方程式(方程式10)和热动力学平衡常数方程式(方程式11)。在该方程式中,copen,i和cclosed分别表示引物3’碱基i处打开和引物3’′封闭的引物-模板复合物的平衡浓度;cc是方程式2中表示的引物-模板复合物浓度;kclosed,i表示引物3’碱基i处打开和引物3’′封闭的结合状态的平衡常数;t是退火温度;r为理想气体常数。在一些实施例中,所涉及的一个或多个聚合酶有3’-5’外切核酸酶活性,并且在引物3′端的一个或多个错配可以在退火过程中去除。当pcr样品中含有一个或多个变体等位基因时,可出现错配。在一些实施例中,所用聚合酶有3’到5’外切核酸酶活性,引物3’端结合的稳定性是通过在引物的3′端具有至多1、2、3或多个核苷酸开放的引物-模板复合物的分数来测量的。计算的准确公式来自于热动力学的技术人员,并遵循本公开的指导。允许开放的确切长度可由实验的特定的聚合酶和特定的退火条件包括退火时间来确定。所公开的方法,使得预测引物3’端结合稳定性的现有技术的方法和工具有了显著改善。所公开的方法对于在实际反应条件下引物3'端结合其模板的分数提供了一种定量预测。相比较而言,现有技术的引物设计方法和工具提供经验记分,惩罚3’端涉及长的a和t的引物序列。在这方面,现有技术的方法是定性的,不提供定量预测的引物性能。图14说明了引物和模板折叠对引物模板结合的影响。该图显示了三个平行反应,包括松弛引物p1401转变为折叠结构p’1402,松弛模板t1402转变为折叠结构t’1412和引物p1401和模板t1402杂交为引物-模板复合物pt1420。kpf、kbf和kpt分别为折叠和杂交反应的平衡常数。在平衡状态下,三个反应用方程式13,14和15表示。质量平衡用方程式16和17表示。三个反应的热动力学平衡常数用方程式18、19和20表示。cp0=cp+cp′+cpt方程式16ct0=ct+ct′+cpt方程式17在该方程式中,cp,ct,cp′和ct′分别是在松弛和折叠状态的引物和模板的平衡浓度;cpt是杂交产物的平衡浓度;和分别是引物和模板的起始浓度;δgpf是特定区段内引物的折叠自由能;δgtf是引发区段内模板的折叠自由能;t是退火温度;r为理想气体常数。在一些实施例中,折叠自由能使用最近邻法计算(见j.santaluciajr.etal.(2004)“thethermodynamicsofdnastructuralmotifs”annu.rev.biophys.biomol.struct.33:415–440)。在一些实施例中,折叠自由能使用计算软件包如unafold计算((n.markhamandm.zuker(2008)unafold:softwarefornucleicacidfoldingandhybridization.inkeith,j.m.,editor,bioinformatics,volumeii.structure,functionandapplications,number453inmethodsinmolecularbiology,chapter1,pages3–31.humanapress,totowa,nj.isbn978-1-60327-428-9)。在大多数应用情况下,起始引物浓度是显着高于起始模板浓度,杂交产物的浓度总是小于起始模板浓度,如方程式20中所示。结合方程式13到17和方程式21的应用条件,我们得到了方程式22的fa,fold,显示的是在竞争性引物和模板折叠反应存在的情况下下,模板被引物杂交的分数。cp0>>ct0>cpt方程式21所公开的方法,对现有技术折叠对引物-模板结合的影响的方法和工具进行了显着的改善。所公开的方法提供了在竞争性引物和模板折叠反应存在的情况下,模板被引物杂交的分数的定量预测。相比较而言,现有技术的引物设计方法和工具提供经验记分,惩罚具有稳定折叠(负折叠自由能)的引物序列。在这方面,现有技术的方法是定性的,不提供定量预测的引物性能。在一些实施例中,对参考以及所有可用的变体等位基因进行方程式13到22的计算。挑选最低fa,fold,作为最坏的情况下候选引物的模板缔合分数。引物的性能是由最坏的情况下的引发效率预测,该引发效率由方程式22的最低fa,fold和方程式12的最低fclose相乘得到。在一些实施例中,为了节省计算时间,方程式22是适用于参考等位基因,方程式7和方程式12适用于参考以及变体等位基因。引物的性能,根据方程式23计算,结合fa,fold,参考模板缔合分数fa,ref,最坏的情况下模板缔合分数fa,min,和最坏的情况下引物3′封闭的分数fclose,min估测。这就完成了常规引物的设计和性能预测。通过移动引物3’位置到参考引物结合模板上另一个预定的位置,重复下一个引物设计和性能预测,直到完成所有预定位置的过程。在一些实施例中,以平铺(tiling)模式布置所述预定位置。在一些实施例中,平铺的增量是参考引物结合模板上的1,2,3,4或多个核苷酸。在一些实施例中,平铺形成在参考引物结合模板的正、负链。在一些实施例中,在预定位置设计的引物包括与预先确定的通用引物(通用引物1或通用引物2)的通用区段互补的通用区段。在一些实施例中,设计了2组引物,每组覆盖所有预定位置,但一组包括与通用引物1的通用区段互补的通用区段,其他组包括与通用引物2的通用区段互补的通用区段。如前所述,ω引物包括5p臂,环和3p臂。环序列基本上由相应的通用引物3’区确定,并提供作为预定参数的一部分。在一些实施例中,引物5p臂的功能是作为锚定引物提供与模板序列的稳定结合,而3p臂检查结合的特异性,将环引入到延伸产物中。在示例性实施例中,从3p臂开始设计引物。类似于常规引物设计,引物的3’位置固定在参考引物结合模板的预定位置,3p臂初始轨迹长度的设置在一个预定值。在一些实施例中,预先确定的初始轨迹长度为5、6、7、8、9或更多。为了计算,假设ω引物通过5p臂已经结合到其模板。适当长度的3p臂足以克服环的积极的自由能和具有足够的结合系数结合到其模板,这是理想的结果。为方便起见,3p臂与模板的结合被称为3p臂关闭。3p臂开放和封闭的平衡反应如方程式24所示,这是一级反应。方程式25显示具有3p臂打开的引物-模板复合物的自由能。末端自由能δgterminal涉及5p臂3’末端的悬挂基序。δg5parm,stack是5p臂的最邻近的叠加自由能。方程式26显示具有3p臂关闭的引物-模板复合物的自由能。δgloop是环的自由能。δg3parm,stack是3p臂的最邻近的叠加自由能。方程式27显示的是3p臂打开和关闭状态之间的自由能差。方程式25、26和27中所涉及的所有基序的自由能计算由santalucia详细描述(j.santaluciajr.etal.(2004)“thethermodynamicsofdnastructuralmotifs”annu.rev.biophys.biomol.struct.33:415–440)。方程式28表示方程式24所示的反应的平衡条件。方程式29是质量平衡方程式。方程式30表示方程式24所示反应的热动力学平衡常数。δg3parm,open=δgterminal+δg5parm,stack方程式25δg3parm,close=δgloop+δg3parm,stack+δg5parm,stack方程式26δδg=δg3parm,close-δg3parm,open=δgloop+δg3parm,stack-δgterminal方程式27cpt=c3parm,open+c3parm,close方程式29在该方程式中,cpt,c3parm,close,和c3parm,open分别表示引物-模板复合物、3p臂关闭的引物-模板复合物和3p臂打开引物-模板复合物的浓度;t是退火温度;r为理想气体常数。通过结合方程式28,29和30,推导出方程式31,其表示具有3p臂结合到模板的ω引物的分数。从方程式31得到的f3parm,close值,然后与预定的阈值f3parm,close,thr相比。在一些实施例中,f3parm,close,thr至少是0.90,0.91,0.92,0.93,0.94,0.95,0.97,0.98,0.99或更多。在一些实施例中,fa,thr是约从0.90到0.99,0.91到0.99,0.92到0.99,0.93到0.99,0.94到0.99,0.95到0.99,0.97到0.99,0.98到0.99。如果f3parm,close比f3parm,close,thr小,可以通过按照1或预定递增数量来增加3p臂的长度,重复上述计算直到f3parm,close接近f3parm,close,thr。接下来,确定5p臂长度。出于示范的目的,凸环(图1c中以122显示)用于我们的ω引物。在凸环的ω结构,5p臂的3’端位置设置为紧靠近3p臂的5’端。出于计算目的,将5p臂看作分离的结合序列。5p臂的长度以类似于常规引物特异性区段的长度得到。衍生的5p臂满足产生预定阈值模板缔合分数的要求。在一些实施例中,该阈值是至少0.90,0.91,0.92,0.93,0.94,0.95,0.97,0.98,0.99或更多。在一些实施例中,fa,thr是约从0.90到0.99,0.91到0.99,0.92到0.99,0.93到0.99,0.94到0.99,0.95到0.99,0.97到0.99,0.98到0.99。通过推导3p臂和5p臂加上预定环序列,我们有完整的候选ω引物序列,用于进一步评估。本公开的一个显著的方面是设计具有宽容变体的引物,以实现对一般群体样品的加强性能。在实施例中,将设计应用于ω引物。宽容变体的ω引物的3p臂以上述同样的方式衍生而来。宽容变体的ω引物5p臂通过对变体等位基因进行方程式1到7的迭代计算,增加5p臂的长度直到阈值模板的缔合分数满足要求得到。这迫使5p臂长度增加从而保持与模板充分结合,即使当引发区存在一个或多个变体。在一些实施例中,计算中使用预定的最大长度以限制5p臂长度在限值内。在一些实施例中,预确定的最大长度至少为40、50、60或更多。在一些实施例中,计算中包括的变体限于snp变体。一般群体中的snp变体的频率是目前所有类型的变体中最高。宽容snp的引物设计可以显著扩大基因组序列中引物放置的可用区域。在一些实施例中,在常规或任何其他类型的引物或探针的设计中应用宽容变体引物的原则。ω引物性能的预测类似于上述常规引物的预测。在一些实施例中,对ω引物的3′端结合稳定性的预测是利用方程式8至12进行的。这产生最坏情况下3’封闭引物分数f3pclose,min。在一些实施例中,使用方程式13到22预测折叠对5p臂结合模板的影响。这就产生了在折叠完成存在的情况下,结合5p臂的模板缔合分数fa,5parm,fold。在一些实施例中,用方程式1到7计算模板结合整个ω引物的模板缔合分数fa,omega。在一些实施例中,用方程式13到22进行预测折叠对模板结合整个ω引物的影响fa,omega,fold。在一些实施例中,更多的考虑预测折叠对3p臂结合的影响。图15示意性地表示出了3p臂结合到模板与折叠的竞争反应。在该示例性说明中,具有与模板1502结合的其5p臂1503的ω引物1501与折叠的模板1512、引物1521、折叠的引物1521、都折叠的引物1531和模板1532、以及期望形式的结合模板1542的3p臂1545的亚稳态处于平衡。这些反应用4个独立的平衡方程式32到35表示。质量平衡用方程式36表示。热动力学平衡常数由方程式37到40提供。cpt=copen+cfold,tpl+cfold,prm+cfold,both+cbind方程式36在该方程式中,cpt,copen,cfold,tpl,cfold,prm,cfold,both,和cbind分别表示5p臂结合到模板的所有状态下引物-模板复合物的浓度,非折叠状态下引物和模板打开状态的引物-模板复合物,模板折叠的引物-模板复合物,引物折叠的引物-模板复合物,模板和引物都折叠的的引物-模板复合物,3p臂结合到模板的引物-模板复合物;δδg3parmclose表示当5p臂结合到模板时,3p臂闭合和3p臂打开状态下自由能差值,该值由方程式27提供;t为退火温度;r为理想气体常数。结合方程式32到36,衍生出方程式41,其表示在存在引物和模板折叠反应竞争的情况下,具有3p臂结合到模板的ω引物的分数。在一个实施例中,ω引物性能由最坏的情况下结合5p臂的变体模板的模板缔合分数fa,5parm,min、结合5p臂的参考模板的模板缔合分数fa,5parm,ref、最坏的情况下引物3’闭合的分数f3pclose,min、存在折叠竞争情况下,结合5p臂的参考等位基因模板的模板缔合分数fa,5parm,fold,和在存在引物和模板折叠反应竞争的情况下,具有3p臂结合到模板的ω引物分数f3parmclose,fold。方程式42为ω引物性能的一个示范性的预测。在一些实施例中,出于计算时间,计算精度、5p臂,3p臂和环区段功能的具体设计和具体应用的考虑,与5p臂结合相关的模板缔合分数(fa,5parm,min、fa,5parm,ref和fa,5parm,fold)可以被相应的与整个ω引物结合相关的模板缔合分数取代。有关整个ω引物结合的模板缔合分数的计算已在上面描述。通过移动引物3’位置到参考引物结合模板上另一个预定的位置上,可以重复下一个ω引物的设计和性能预测,直到完成所有预定位置的过程,方式与上述常规引物的描述一样。在一些实施例中,其中特异性引物的制备包括pcr扩增,引物的性能预测,包括引物(常规,ω引物,以及其他类型)的pcr模板900,1000和1100的扩增效率。在一些实施例中,计算模板侧翼区段901、905、1001、1003、1101、和1103中模板的折叠对pcr翻译中引发效率的影响。在一些实施例中,使用方程式13到22进行计算。引物设计过程的步骤3至5寻找引物偏离靶的位置,并提供在偏离靶位置,引物延伸效率的定量预测。在步骤3中,所有候选引物都与参考基因组数据库或任何序列数据库比对,基本上代表候选引物将要被应用的样品中所涉及的完整dna序列组。在某些实施例中,使用blast或一个或多个blast衍生软件进行比对。为方便起见,比对结果被标记为命中。从总命中中删除一个靶命中。靶向命中是引物被设计结合的序列位点。其余的命中是偏离靶的命中。保存比对位置,查询序列的比对部分和主序列的比对部分,用于偏离靶的结合系数计算使用。引物序列是比对的查询序列,和数据库序列是主序列。在一些实施例中,使用blastn。在一些实施例中,以这样一种方式设置blast,允许错配和空位。在一些实施例中,blast空位开放、空位延伸、惩罚和奖励使用默认值。在一些实施例中,以这样一种方式设置blast,允许错配和空位。在一些实施例中,使用合理的小blast字段长度(word_size)保证偏离靶搜索的灵敏度。在一些实施例中,blast字段长度设置为6,7,8,9,10,11,12,13,14,15或更多。该字段长度的选择是基于计算时间,可用计算机内存,和预期比对搜索灵敏度之间的平衡。blast是最流行的和最基本的比对工具,它的使用是生物信息学的技术人员所熟悉的。在一些实施例中,其中对ω引物进行偏离靶的搜索,除了原始整个ω引物序列的对比外,增加了需要的比对操作。增加的比对捕获任何偏离靶的位点,该位点可以与ω引物杂交形成稳定的环结构。blast是局部比对工具,可能会错过涉及大空位的这种类型的位点。在一些实施例中,通过与目的序列数据库比对组合的3p臂和5p臂序列形成额外的比对。在比对结果中,当查询序列的比对部分包括5p臂和3p臂的连接,插入相应的环序列到查询序列的比对部分连接,以产生恢复查询序列,并将相同环长度的空位插入到相应主序列的比对部分位点的连接,以产生比对主序列。然后通过去除整个ω引物比对结果和组合的5p臂和3p臂序列比对结果之间多余的比对命中,编译比对结果。在一些实施例中,使用全局比对工具对整个ω引物序列的序列对和相应的扩大区域做两两比对。在一些实施例中,全局比对工具是基于needleman–wunsch算法编写(s.needlemanelal.(1970)"ageneralmethodapplicabletothesearchforsimilaritiesintheaminoacidsequenceoftwoproteins"journalofmolecularbiology48,443–53)。比对结果将有适当的空位。引物设计的步骤4是构建引物/偏离靶的结合对序列,为计算结合系数做准备。通过扩展查询序列和主序列比对部分两端预定数量的核苷酸来构造配对序列。在某些实施例中,预定数量是0、1、2、3或更多。扩大比对部分的目的是在结合对序列两端产生突出端,这样下面结合计算可以通过包括突出端叠加能量更准确地完成(j.santaluciajr.etal.(2004)“thethermodynamicsofdnastructuralmotifs”annu.rev.biophys.biomol.struct.33:415–440)。引物设计的步骤5是计算引物/偏离靶的结合特性。使用上述与靶向模板与引物结合相同的方法,计算引物和偏离靶序列之间的结合自由能。从自由能,我们在特异性引物退火条件下使用方程式1到7得到偏离靶序列的结合系数。另一个重要的偏离靶结合特性是结合区域末端和引物3’端之间的距离也被称为距离突出端长度。当突出端长度小于阈值长度,认为偏离靶引物是可延伸的。在一些实施例中,通过所用的聚合酶确定阈值长度。在一些实施例中,当聚合酶没有3’-5’外切核酸酶活性,阈值的长度是0。在一些实施例中,当聚合酶具有3’-5’外切核酸酶活性,阈值的长度至少是0,1,2,3或者更多个核苷酸。在一些实施例中,保存整组引物/偏离靶的结合特性,包括位置,结合系数和可延伸性,当结合系数超过阈值结合系数值时使用。在一些实施例中,可延伸的偏离靶结合的阈值结合系数值是0.05,0.01,0.005或更少。在一些实施例中,非可延伸的偏离靶结合的阈值结合系数的值是0.75,0.5,0.25,0.1或更少。阈值结合系数的值是由所需的引物特异性,计算时间,计算机内存大小,和计算机存储大小来确定。引物设计的步骤6是计算候选特异性引物和通用引物之间的交叉杂交结合。在一些实施例中,通用引物与所有候选引物比对。然后,比对的结果以步骤5中所描述的相同方式被用来计算结合系数。在一些实施例中,交叉杂交自由能使用计算软件包如unafold计算(n.markhamandm.zuker(2008)unafold:softwarefornucleicacidfoldingandhybridization.inkeith,j.m.,editor,bioinformatics,volumeii.structure,functionandapplications,number453inmethodsinmolecularbiology,chapter1,pages3–31.humanapress,totowa,nj.isbn978-1-60327-428-9)。对于大多数应用,每个特异性引物的浓度显著小于通用引物的浓度,并且结合系数是通用引物结合杂交的特异性引物的分数。应检查特异性引物和通用引物的引物延伸性。如果可延伸的引物被发现,进一步检查是否延伸产物可以由一个或两个通用引物扩增。如果延伸产物是由通用引物从常规引物的特异性区段或从ω引物的3p臂区段延伸产生,那么这个延伸产物是可扩增的。当结合系数超过阈值结合系数值时,记录候选引物的结合系数、延伸性、和可扩增性。在一些实施例中,非可延伸的交叉杂交结合的阈值结合系数的值是0.25,0.2,0.1或更少。在一些实施例中,可延伸但不可扩增的交叉杂交结合的阈值结合系数值是0.2,0.1,0.05或更少。在一些实施例中,可延伸和可扩增的交叉杂交结合的阈值结合系数的值为10-5、10-6、10-7、10-8或更少。通过保持合理高部分的用于特异性引发用途的特异性引物的考虑来确定非可扩增的交叉杂交结合的阈值结合系数。可扩增的交叉杂交结合的阈值结合系数值是通过考虑特异性引物和接力pcr模板之间的浓度比确定。目标是保持引物二聚体产物的水平低于模板扩增的水平。作为示例性的例子,假设模板浓度为1fm和特异性引物浓度是10pm;可扩增交叉杂交结合的结合系数为10-5会产生约0.1fm引物与引物延伸产物,并会与模板平行扩增。最终,约10%的pcr产物为引物-引物二聚体。当单独候选特异性引物真正被选择时,在计算过程的步骤9中进行特异性引物中交叉杂交结合的计算。引物设计的步骤7是计算所有候选引物的记分。在一些实施例中,除了定量计算步骤2中导出的引发效率外,我们使用几个质量记分来指导从候选引物群中选择最终的引物。在示范性实施例中,质量记分公式化以有利于引物gc含量偏向于0.4和0.6之间,以阻止引物长度超过90,有利于在参考等位基因对比最坏情况下变体等位基因上所获得的高比率的引物长度,以阻止偏离靶的结合,并阻止特异性引物和通用引物之间的交叉杂交结合。以前步骤的计算已经产生了记分计算所需的参数。已经计算的参数包括引物长度、参考等位基因模板的引物长度,最坏情况下变体等位基因模板的引物长度,可延伸和不可延伸的偏离靶结合的结合系数,不可延伸的结合系数,可延伸但不可扩增的结合系数,以及候选特异性引物和通用引物之间可扩增的交叉杂交的结合。记分公式使用线性,非线性,成比例,成反比例,和某些类型的分布曲线,这是做控制,自动化,和传统的pcr引物设计的技术人员所熟悉的。在示范性的实施例中,所有的记分都有最大值1和最小值的0,1是最好的,0是最差的。按照方程式式43,把所有的单个记分结合成一个单一的记分,来表示引物的整体质量。在该方程式中,si是单个记分,包括gc含量,引物长度,参考等位基因与最坏情况下变体等位基因的引物长度比率,特异性引物与通用引物之间的偏离靶结合和交叉杂交结合。在该方程式中,wi是记分si的权重因子。权重因子是一个按经验确定的数,用于调整单个记分在确定整体质量记分的相对重要性。权重因子通常具有最小值0。引物设计的步骤8是建立候选捕获区域的列表。该列表包括捕获区域的起始和终止位置,同时每个候选捕获区域具有特异性引物1和特异性引物2。候选捕获区域的目的是用丰富的特异性引物1和特异性引物2的组合,提供所需靶区域的全部覆盖,这样可以通过选择最佳可能的特异性引物1和特异性引物2的组合,最终选择出捕获区域。“候选捕获区域”和“捕获区域”可互换地使用。在一些实施例中,以平铺(tiling)模式设置捕获区域。在一些实施例中,捕获区域的长度是预先确定的。在一些实施例中,根据应用确定预定区域长度。在示例性实施例中,所捕获的序列用于高通量测序。根据测序读长,确定捕获区域的长度。测序读长是测序仪器单次读取通道报告的核苷酸的数量。作为示例性的例子,测序运行包括2个通道,每个通道读长为150nt(核苷酸)。两个通道从相反的两端读取靶序列。总测序长度为300个核苷酸。然后,为了读取整个捕获区域,在2次读取通道之间有一定的重叠,280个核苷酸可以被选择作为最大捕获区域长度。为选择最佳可能的特异性引物,在该区域长度上保持一定的灵活性是非常有用的。140个核苷酸被选为最小捕获区域长度。处理超过最大捕获区域长度的长重复屏蔽部分的情况能力也很有用。重复屏蔽序列往往可以有非常大的数量发生在基因组中,并常常被引物设计所屏蔽。为了充分覆盖所需靶区域,当平铺过程运行到重复屏蔽区域,扩展最大捕获区域长度。为了示例性目的,我们设置了扩展的最大捕获区域长度在600。在一些实施例中,靶的平铺过程从挑选正链最左(或最低位置)的候选特异性引物1开始,然后选择位于最小捕获区域长度上游的负链的候选特异性引物2。确保所需靶区域中至少有一部分是在两个特异性引物之间。候选扩增子从候选捕获序列的pcr扩增产生。候选的扩增子的质量记分是两特异性引物的质量记分的产物。在一些实施例中,计算包括但不限于gc含量、折叠自由能和候选捕获区域长度的其它质量记分。通过上述步骤7的下面的教导,可以得出以分数公式。扩增子的整体质量记分计算与方程式43相同。添加捕获区域的开始和终止位置,特异性引物1和特异性引物2的信息,和候选捕获区域列表的扩增子记分。重复上述过程,不断增加捕获区域长度,直到达到最大捕获区域长度。在一些实施例中,增量为1、2、3、4、或更多。然后从下一个正链的最左的候选特异性引物1开始,重复上述过程,直到平铺达到最右的可用候选特异性引物。在一些实施例中,进行平铺附加轮循环是从正链最左边的候选特异性引物2,负链特异性引物1和负链特异性引物2开始,每个引物与相对链的其它特异性引物相匹配。引物设计的步骤9是选择扩增子序列覆盖所有所需的靶序列。在这部分的计算中,扩增子,包括相应的特异性引物的最后选择是在候选捕获区域列表上的所有可用候选扩增子中进行。在一些实施例中,最后的扩增子以平铺的方式被选择。在一些实施例中,平铺过程被应用于一次一个靶序列。在一些实施例中,靶序列的平铺选择开始于覆盖靶序列的起始位置的候选扩增子组。在该组中,按上述步骤6相同的方式,计算候选特异性引物对和每个候选特异性引物自身之间的交叉杂交结合。以上述步骤7中所描述的相同的方式计算交叉杂交结合的记分。下一步,通过候选特异性引物对偏离靶的序列的捕获进行预测。这是通过查找步骤5中的引物/偏离靶结合特性的数据,并决定是否有可测量的偏离靶序列可以通过候选特异性引物捕获来完成的。当在相同的毗连序列上都有可延伸的偏离靶结合位点的两个特异性引物有位于相对链上彼此面对的3’端时,并彼此足够接近时,他们会产生偏离靶序列。这方面的考虑包括单个特异性引物具有2个或更多个偏离靶结合位点。在一些实施例中,仅当偏离靶结合位点之间的距离低于阈值距离时才考虑偏离靶捕获。在一些实施例中,阈值距离是最多100000,10000,1000或更少。偏离靶产物的相对浓度从两偏离靶结合位点的结合系数产物预测。基于偏离靶产物的相对浓度计算候选扩增子(这是与候选引物相关)的偏离靶记分。在一些实施例中,偏离靶记分被公式化成与偏离靶产物的相对浓度成反比,其记分范围从0到1。由原始质量记分乘以交叉杂交结合记分和偏离靶记分来修改相应扩增子的质量记分。然后,选择具有最高的质量记分的扩增子,作为第一平铺。如果选定的扩增子覆盖靶序列的终止位置,靶序列的选择完成。如果选定的扩增子没有覆盖靶序列的终止位置,继续进行下一个平铺。在一个优选实施例中,相邻的平铺在接合处的一些长度重叠。在一些实施例中,重叠长度至少为1、2、5、10、15、或更多个核苷酸。在一些实施例中,作为输入参数的一部分,预先确定最小重叠长度和最大重叠长度。在一些实施例中,最小重叠长度至少为1、2、5或更多个核苷酸。在一些实施例中,最大重叠长度至少是10,20,30或更多个核苷酸。在一些实施例中,为了避免相邻平铺之间捕获反应(图3、4a、4b、5的第1轮循环和第2轮循环)的干扰,相邻平铺的接力pcr反应在单独的pcr管中进行。在一些实施例中,两个pcr管用于一套完整的靶序列的捕获和扩增,每个管包含用于平铺中每一个其他成员的特异性引物。保存第一平铺扩增子的引物信息到管1引物列表。第二平铺选自候选扩增子组,其在第一平铺的捕获区域的终止位置减去最大重叠长度再减去最小重叠长度范围内有捕获区域开始位置。从这个组中,以上面描述第一平铺同样的方法挑选第二平铺扩增子。保存第二平铺扩增子的引物信息到管2引物列表。如果选定的扩增子覆盖靶序列的终止位置,靶序列的选择完成;如果选定的扩增子没有覆盖靶序列的终止位置,继续进行下一个平铺,直到达到靶序列的末端。当引物已经存在于管1或管2时,预先存在的引物需要被包含在特异性引物交叉杂交结合和偏离靶捕获的计算中。例如,如果按上述过程继续第三平铺,新选择的引物将放置在管1。两种特异性引物交叉杂交结合和偏离靶捕获的计算将包括新的特异性引物对,每个新的特异性引物与自身,和每个新的特异性引物与管1中已有的每个引物之间的结合。在一些实施例中,对含有长同源部分的靶序列的平铺选择进行特殊的考虑。如上所述,在步骤1中已识别了长的同源部分,并在靶数据表中提供信息。在一些实施例中,基于同源部分的一个拷贝选择扩增子,相应的特异性引物用于所有同源部分。这样可以避免了从不同位点,而本质上相同的靶序列复制和/或冲突引物的潜在问题。显然,在上述教导的指引下,对所公开的计算方法进行许多修改和变化是可能的。例如,在一些实施例中,总是去除低质量的引物,以减少以后步骤的计算时间。应用除了上述靶富集用于大规模平行测序,本发明可以有利地用于各种其他应用中。在一些实施例中,该ω引物与接力pcr组合有利地用于实时pcr。每个反应中使用了一对特异性引物和一对通用引物。热循环过程期间,在前两轮循环中,特异性ω引物高特异性和高效率地筛选靶部分。在剩下轮的循环中,通用引物对靶进行扩增。这种方法的优点是无论靶序列是什么,都在扩增循环中使用一对表征很好地的通用引物。期望对靶序列变异的测量依赖性比常规实时pcr减少,在实时pcr中特异性引物负责所有扩增循环。传统方法中成指数地扩大了由于不同特异性引物设计引起的扩增产率差异。通过使用接力pcr,由于不同的特异性引物设计而产生的扩增产率差异被消除。在一些实施例中,常规的特异性引物与接力pcr的组合被应用于实时pcr。在一些实施例中,使用接力pcr制备阵列检测用的样品。每个接力pcr反应中都采用了多个特异性引物和一对通用引物。在一些实施例中,至少有一种通用引物附着有荧光染料,包括但不限于cy3,cy5,alexa3,alexa5,ftic和fam(这里添加更多)。在一些实施例中,至少有一种通用引物连接有共轭配体,包括但不限于生物素,nhs,nh2和cho。荧光染料和配体这里被称为标记。一个或多个标记可以连接到引物分子的一个或多个核苷酸上。标记连接的引物被称为标记引物。在一些实施例中,一对通用引物由一个未标记引物和一个标记引物组成。在一些实施例中,标记引物的浓度高于未标记引物的浓度。标记和未标记引物的摩尔浓度比为至少1,2,5,10或更大。在一些实施例中,使用两轮pcr反应来产生标记的靶样品。在第一轮中,进行接力pcr。用多个特异性引物和一对未标记的通用引物。第一轮pcr产物可选择性地被纯化,以除去残留的引物和酶,并保留双链pcr产物。第二轮是单链pcr反应,涉及一个标记的通用引物和等量的第一轮pcr产物或纯化的第一轮pcr产物。在一些实施例中,该等量至少是第一轮pcr产物的总体积的1/1000,1/500,1/200,1/100,1/50,1/20,1/10或更多。在一些实施例中,该组合物包括可扩增短串联重复序列,单核苷酸多态性,基因,外显子,编码区,外显子组,或其它部分的一个或多个靶特异性引物对。在一些实施例中,模板是由rna样品合成的cdna。因此,在一些实施例中,提供了寡核苷酸引物,其包含具有3’端和5’端的3p臂,环部分和具有3’端和5’端的5p臂,其中5p臂杂交dna模板,3p臂杂交dna模板和提供聚合酶延伸的序列特异性,环部分位于5p臂和3p臂之间,不与dna模板结合。在一些实施例中,dna模板基本上与5p臂和3p臂互补。在一些实施例中,5p臂是10到100个核苷酸的长度,如25到60个核苷酸;和/或3p臂的长度是6到60个核苷酸,如10到20个核苷酸;和/或环部分是12到50个核苷酸的长度,如15到40个核苷酸。在一些实施例中,当结合到dna模板时,3p臂的5’端和5p臂的3’端彼此邻近。在一些实施例中,当与dna模板杂交时,5p臂比3p臂具有较高的结合能(如两或三倍高)。在一些实施例中,该引物包括凸环、发夹环和/或内环。在一些实施例中,该公开提供了一种用于检测使用的杂交结构,包括探针和靶,其中杂交结构具有一个或多个单链环和2个或多个双链体区段,其中每个环路位于双链区段之间。在一些实施例中,该单链环在探针中,包括一个或多个非核苷酸部分。在一些实施例中,该探针包括间隔区。在一些实施例中,杂交结构用于聚合酶延伸。在一些实施例中,杂交结构用于杂交检测。在一些实施例中,环包含12到50个核苷酸。在一些实施例中,提供用于扩增靶核酸的方法。在一些实施例中,该方法包括提供第一特异性引物,第一通用引物,第二通用引物,侧翼的靶片段,聚合酶和核苷酸;进行靶筛选,包括一轮第一热循环程序,该程序包括变性步骤、退火步骤和延伸步骤;以及进行扩增,其包括两轮或多轮第二热循环程序,该程序包括变性步骤、退火步骤和延伸步骤,从而扩增靶核酸。在一些实施例中,该方法包括提供第一特异性引物,第二特异性引物,第一通用引物,第二通用引物,靶核酸,聚合酶和核苷酸;进行靶筛选,包括两轮第一热循环程序,该程序包括变性步骤、退火步骤和延伸步骤;以及进行扩增,包括两轮或多轮第二循环程序,该程序包括变性步骤、退火步骤和延伸步骤,从而扩增靶核酸。在这些方法的一些实施例中,第一特异性引物有3’端和5’端,其中3’端含有第一序列特异性区段和5’端包含第一通用区段;和/或第二特异引物有3’端和5’端,其中,3’端含有第二序列特异性区段和5’端包含第二通用区段;和/或第一和第二特异性引物的浓度低于第一和第二通用引物500倍,和/或第一和第二特异性引物的浓度约约从0.0001nm到5nm;和/或第一和第二通用引物的浓度约从200nm到5000nm;和/或第一和第二特异性引物的浓度小于1nm;和/或第一和第二通用引物的浓度大于200nm;和/或第一个特异性引物是ω引物,并且其中第二个特异性引物是ω引物;和/或第一热循环程序的退火时间约从30分钟到4小时;和/或第一热循环程序的退火温度约从60℃至75℃;和/或第一热循环程序的退火温度约从60℃至72℃;和/或第一热循环程序的退火温度约从65℃至72℃;和/或第二热循环程序有从10-50个循环,如20-40个循环,和/或退火温度在聚合酶的聚合酶活性的峰值10℃以内;和/或聚合酶是无链置换活性和5’-3’核酸酶活性的聚合酶;和/或聚合酶选自下列组:phusion热启动flexdna聚合酶和热启动高保真dna聚合酶。在一些实施例中,提供用于扩增2个或多个不同靶核酸的方法。在一些实施例中,该方法包括提供一套特异性引物对,其中每对包括针对特定靶核酸的第一和第二特异性引物,第一通用引物,第二通用引物和一套靶核酸,聚合酶和核苷酸;进行两轮第一热循环程序,该程序包括变性步骤,退火步骤和延伸步骤;进行两轮或多轮第二热循环程序,该程序包括变性步骤、退火步骤和延伸步骤,从而扩增靶核酸。在一些这样的实施例中,第一和第二特异性引物的浓度低于第一和第二通用引物500倍,和/或第一和第二特异性引物的浓度约从0.0001nm到5nm;和/或第一和第二通用引物的浓度约约从200nm到5000nm;和/或第一和第二特异性引物的浓度小于1nm;和/或第一和第二通用引物的浓度大于500nm;和/或第一循环程序的退火时间约从30分钟到4小时,和/或第一和第二特异性引物是ω引物或第一和第二特异性引物是常规特异性引物。在一些实施例中,提供用于纯化pcr产物的方法。在一些实施例中,该方法包括向pcr反应混合物添加组分,该组分包含靶序列、第一通用引物和第二通用引物,dna片段、聚合酶、pcr缓冲液,其中靶序列侧翼有引发片段,其与第一通用引物和第二通用引物是相同或互补的,不包含引发区段的片段,以及第二通用引物包括引发区段、修饰基因区段和标记段;探针接枝珠,其中探针具有与标记区段的序列基本上互补的序列,和便于通过杂交通过珠捕获pcr产物。在这种方法的一些实施例中,修饰基因区段选自下列群组:一个或更多的c3烷基间隔区,一个或多个乙二醇间隔区,一个或多个光裂解的间隔区,一个或多个1’,2’-双脱氧核糖、一个或多个脱氧尿苷或它们的组合。在一些实施例中,标记段包括至少一个结合部分。在一些实施例中,结合部分可以是生物素。在一些实施例中,标记区段可以包含寡核苷酸和结合部分。在一些实施例中,结合部分可连接到标记区段寡核苷酸的5'端。在一些实施例中,提供用于产生表面簇的方法。在一些实施例中,该方法可包括用第一通用引物和第二通用引物扩增靶序列,其中第一通用引物包括引发区段和第二通用引物包括引发区段,修饰基因区段和标记区段,以产生包含单链标记的pcr产物,其中,pcr产物包括第一和第二链,其中第二链连接到单链标记;提供基底,所述基底包括探针、第一表面引物,第二表面引物;将pcr产物和引导应用到基底上,使得pcr产物、引导与探针杂交,以在基底表面上上产生pcr产物/引导/探针复合物;连接pcr产物与探针从而耦合pcr产物;冲洗基底去除pcr产物的第一链;以及延伸第一表面引物和第二表面引物,从而形成表面簇。在一些这样的实施例中,探针,第一表面引物,以及第二表面引物附着于基底。在一些实施例中,探针、第一表面引物和第二表面引物还包括间隔区,通过该间隔区,该探针、第一表面引物以及第二表面引物可连接到基底表面。在本文所述方法的一些实施例中,通用引物可含有尾区段和通用区段;和/或特异性引物具有特异性区段和通用区段。在一些实施例中,这里描述的引物可以有通用区段和特异性区段,其中特异性区段是由3p臂5p臂组成,其中环是由通用区段组成。在一些实施例中,提供了一种设计pcr引物的方法。在一些实施例中,该方法可包括确定引物长度,以产生足够的模板缔合系数;确定引物3’端结合系数;确定折叠效应存在情况下模板缔合系数;通过结合变体等位基因的缔合系数确定引发效率。为了更好地了解本发明和其许多优点,将通过下面的实施例以示例的方式给出。实施例实施例i——常规和接力pcr的比较λdna(来自neb,lpswich,ma)作为模板,设计两个常规特异性引物和两个通用引物,使用phusionhotstartflex聚合酶mastermix(来自neb,lpswich,ma)。下面列出各个反应混合物的组分。管1中的反应是常规pcr。管2和3中反应是利用不同特异性引物浓度的接力pcr反应。表1:反应混合物组分单位管1管2管3特异性引物(λprm1和λprm2)nm(每种引物)50050.5通用引物(comprm1和comprm2)nm(每种引物)500500λdna模板fm101010phusionhotstartflex2×mastermixx111总体积μl252525热循环反应在热循环仪dnaenginetetrad(来自bio-rad,hercules,ca)中进行。管1中的常规pcr使用的温度程序如下表。表2:pcr温度程序步骤温度(℃)时间活化19830秒变性29815秒退火36030秒延伸47230秒转到2,1个循环5变性69815秒延伸77230秒转到6,25个循环8延伸97210分钟保持104永远管2和3中的接力pcr反应温度程序如下表。表3:接力pcr温度程序特异和通用引物序列如下表。所有实验中用到的寡核苷酸序列由lcsciences,houston,tx提供。除了特别描述的,所有寡核苷酸序列使用传统合成方法在cpg(可控孔径的玻璃)基板(l.j.mcbrideetal.(1983)““aninvestigationofseveraldeoxynucleosidephosphoramiditesusefulforsynthesizingdeoxyoligonucleotides”tetrahedronletters,24:245248)上合成。表4:引物序列列表pcr产物利用3%琼脂糖凝胶电泳分析。琼脂糖凝胶通过1.2g琼脂糖溶解在40ml1×tae中,根据琼脂糖制造商的说明转化为凝胶厚板(grandisland,ny)来制备。12×7-mm梳子用于制造加样孔。凝胶加样,来自每个pcr产物管(25μl)的1μlpcr产物溶液与1μl6×蓝色凝胶加样缓冲液和4μltae在pcr管中混合。混合物充分混合,旋转,加样到凝胶加样孔中。70v电泳1小时20分钟。这种凝胶厚板按照生产商说明用sybrgold染色(grandisland,ny)。图16是实验的琼脂糖凝胶电泳图像。泳道1-3对应的管是1-3产物。泳道l是50bp的序列梯走胶。在所有三个管中都能获得预期大小的pcr产物。管1中的常规pcr包括两种特异性引物和219bp的预期产物大小。在这个反应中,两种常规引物浓度为500nm,退火时间30sec足以产生预期量的预期产物,如在图16的泳道1中显示。在最开始的两个循环中,60℃的退火温度由特异性引物的靶特异性部分的tm确定。在剩余的循环中,使用在72℃的组合的退火延伸步骤。温度是由整体特异性引物的tm值确定的。在管2和3中的多接力pcr包括两个特异性引物和两个通用引物。预期产物大小为290bp。管2和管3中使用特异性引物的浓度分别为5nm和0.5nm低浓度。在开始的两个循环中使用超长的1小时的延长退火时间,从而允许低浓度特异性引物和相应模板的杂交。在最开始的两个循环中,60℃的退火温度由特异性引物的靶特异性部分确定。设计循环3和4是为了添加通用引物侧翼到循环1和2产生的靶序列。循环3和4的60℃的退火温度由通用引物的通用区段的tm值确定。在剩余的循环中,在72℃使用结合的退火延伸步骤。温度是由整体通用引物的tm值确定。在这个反应中,使用常规通用引物浓度为500nm。从循环3到最后的循环使用30sec的短退火时间用于扩增。在图14的泳道2和3中显示凝胶电泳图像中观察到的预期量的预期产物。同时也进行了对照试验,第一个对照试验包括两种分别500nm的通用引物(comprm1和comprm2),λdna10fm以及phusionhotstartflex聚合酶mastermix(来自于neb,ipswich,ma)。第二个对照试验包括两个分别50nm的特异性引物(λprm1和λprm2),两个分别500nm的通用引物(comprm1和comprm2),没有λdna和phusionhotstartflex聚合酶mastermix(来自于neb,ipswich,ma)。两组对照实验使用如上所示接力pcr温度程序进行。在两个反应中都没有观察到产物。实施例ii——接力pcr利用ω引物扩增人类基因组dna人类dna用作模板,使用两个常规的特异性引物和两个通用引物,phusionhotstartflex聚合酶mastermix(来自于neb,ipswich,ma)被利用。六对特异性引物用于分别扩增六个靶序列。一对通用引物结合每对特异性引物使用。加上一个非特异性引物对照的总共六个扩增反应分别在六管中进行。反应混合物的组分如下所示。表5:反应混合物的组分单位管6管7特异性引物(specprm1和specprm2)nm(每种引物)1通用引物(comprm1和comprm2)nm(每种引物)500500人类基因组dna模板fm22phusionhotstartflex2×mastermixx11总体积μl2525热循环反应利用以下的温度程序在热循环仪dnaenginetetrad(来自bio-rad,hercules,ca)中进行。表6:接力pcr温度程序步骤温度(℃)时间活化1985分钟变性29815秒特异性引物退火3652小时延伸1468120秒延伸2572120秒转到2,1个循环6变性79815秒起始通用引物退火86030秒延伸1968120秒延伸21072120秒转到7,1个循环11变性129815秒延伸11368120秒延伸21472120秒转到12,25个循环15延伸167210分钟保持174永远特异性引物序列信息如下表:表7:特异性引物序列信息索引引物名称反应管靶染色体靶链引物链靶起始/终止靶长度扩增子长度1tp53_31_59_tile03o11chr17+-75786152tp53_31_59_tile03_o21chr17++75784391762933tp53_31_59_tile01_o12chr17+-75783534tp53_31_59_tile01_o22chr17++75781621913085pik3ca12_tile01_o13chr3+-1789389826pik3ca12_tile01_o23chr3++1789388091732907kras1_tile01_o14chr12-+253784058kras1_tile01_o24chr12--253785951903079apc1_tile01_o15chr5-+11217387110apc1_tile01_o25chr5--11217404217128811apc2_tile01_o16chr5-+11217455712apc2_tile01_o26chr5--112174730173290表8:引物序列列表使用的通用引物与实验i中相同。pcr产物分析采用与实验i中相同的琼脂糖凝胶电泳方法。图16就是凝胶图。泳道1到泳道6分别是六个单独pcr反应对应的产物。泳道7是非特异性引物对照走胶结果。泳道l是dna序列梯用来显示对应标准参照物的大小(碱基对或bp)。在六个管中都获得了预期大小的pcr产物。下表显示产物条带在凝胶图上的相对信号。这个相对信号的标准偏差是0.191。利用分析软件(来自mediacybemetics,rockville,md)提取凝胶图像中的凝胶信号值。相对信号是提取信号除以信号值得到。在泳道7的非特异性引物实验中没有检测到pcr产物。表9:产物条带的相对信号实施例iii——多重pcr在一个管中扩增实施例ii的所有六个靶与实施例ii中同样的六对ω引物和一对通用引物被使用,反应混合物的组分如下表。表10:反应混合物组分单位管1特异性引物(12个引物)nm(每种引物)1通用引物(comprm1,comprm2)nm(每种引物)500人类基因组dna模板fm2phusionhotstartflex2×mastermixx1总体积μl25热循环反应利用以下的温度程序在热循环仪dnaenginetetrad(来自bio-rad,hercules,ca)中进行。表11:接力pcr温度程序pcr产物分析采用与实验i中相同的琼脂糖凝胶电泳方法。图18就是凝胶图。泳道1是多重pcr的产物。泳道l是dna梯序列用来显示对应标准参照物的大小(碱基对或bp)。pcr产物的大小分布在预期的288到308bp。多重pcr产物使用hiseq2000(来自illumina,sandiego,ca)通过平行测序分析。所有六个预期扩增子都生成了测序读取。下表显示每个靶序列的扩增子的读取数分布。在表中,读取分数通过每个目标序列的读取数除以总读取数来计算。相对读取分数通过读取分数除以读取分数的平均值来计算。所有6个扩增子都有平均读取数的20%以上的读取数。相对读取分数的标准偏差是0.512。图18b是6个预期扩增子的测序读取数分布的散布图。表12:测序得到的每个扩增子的读取数分布实施例iv——利用ω引物的多重pcr在一管中扩增44个靶在一个反应管中进行利用ω引物的多重接力pcr,以捕获和扩增人类基因组dna中44个靶。pcr产物的扩增子分布通过hiseq2000(来自illumina,sandiego,ca)测序仪测序来获得。根据公开的引物设计和计算方法,88个ω引物被设计用来捕获人类基因组的44个特异性靶区域。基因组组装版本grch37/hg19用于靶/引物设计。在下面表中列出了捕获区域的信息。在表中,我们称包括ω引物3p臂区段的捕获区域为“探针”(prb)。表的最后两列列出了成对引物的索引。ω特异性引物序列根据本公开的设计方法设计,通用引物与实验i中通用引物相同。表13:捕获区域列表反应混合物组分如下表。表14:反应混合物组分单位管a管b管c特异性引物(88个引物)nm(每种引物)10.20.04通用引物(comprm1和comprm2)nm(每种引物)500500500人类基因组dna模板fm222phusionhotstartflex2×mastermixx111总体积μl252525热循环反应利用以下的温度程序在热循环仪dnaenginetetrad(来自bio-rad,hercules,ca)中进行。表15:接力pcr温度程序步骤温度(℃)时间活化1985分钟变性29815秒特异性引物退火3652小时延伸146860秒延伸257260秒转到2,1个循环6变性79815秒最初通用引物退火86030秒延伸196860秒延伸2107260秒转到7,1个循环11变性129815秒延伸1136860秒延伸2147260秒转到12,25个循环15延伸167210分钟保持174永远通过利用hiseq2000(来自illumina,sandiego,ca)平行测序分析多重pcr产物。图19显示扩增子读取数分布的测序测量结果。图a、b和c、绘制了分别利用1nm、0.2nm和0.04nm浓度的特异性ω引物获得的结果。所有44个设计的靶区域产生的扩增子在测序结果中观察到。来自所有三个特异性ω引物浓度的pcr产物中超过95%的扩增子有在平均读取数20%以上的读取数。实施例v——接力pcr和退火时间进行实验以揭示依靠第一阶段各自5nm和1nm的特异性引物浓度的特异性引物退火时间的多重pcr产率。反应混合物的组成成分如下所示。表16:反应混合物组分单位管1-5管6-10特异性引物(apc1_r1,apc1_r2)nm(每种引物)51通用引物(comprm1和comprm2)nm(每种引物)500500人类基因组dna模板fm22phusionhotstartflex2×mastermixx11总体积μl2525特异性引物退火时间min10-24010-240特异性引物序列如下。通用引物与实验i中所用通用引物相同。表17:引物序列列表引物名称引物序列5′到3′apc1_r1gttcagagttctacagtccgacgatcgagagaacgcggaattggtctaggcaseqid17apc1_r2ccttggcacccgagaattccaagtggtagacccagaacttctgtcttcctseqid18热循环反应利用以下的温度程序在热循环仪dnaenginetetrad(来自bio-rad,hercules,ca)中进行。表18:接力pcr温度程序pcr产物分析采用与实验i中相同的琼脂糖凝胶电泳方法。在所有10管中,预期pcr产物大小288bp已获得。下表显示胶图中所提取的产物条带的信号值和相对信号值。利用分析仪软件(来自mediacybernetics,rockville,md)提取凝胶图像中的凝胶信号值。相对信号值是指在同一特异性引物浓度内相应的信号值除以最大信号值得到的。从数据中可以看出在5nm的高特异性引物浓度时,在10分钟的短时特异性引物退火时间内,相对信号值迅速升高到0.7高的相对信号。但是,在1nm的低特异性引物浓度时,需要显著延长的特异性引物退火时间60分钟或更长使相对信号值接近于1。表19:产品条带信号实施例vi–利用微阵列合成的特异引物前体的多重接力pcr我们进行实验以实施利用微阵列合成的特异引物前体的多重接力pcr实验。这个实验的方法在本公开的图9的示例中已经有介绍。根据本公开的设计方法,该实验设计了捕获人基因组中102个特异靶区域的一组204个ω引物前体序列,所有的ω引物前体序列拥有相同的5’和3’侧翼区段用于pcr扩增使用。下面的表格列出了2个示例的ω引物前体序列(204个中的2个)和2个制备引物序列。如本公开中之前所述,制备引物prepprm2在其3’末端有du。204个ω引物前体序列就是使用微阵列合成方法合成的,而2个制备引物是使用传统的寡核苷酸合成方法合成。所有的204个ω引物前体序列都是使用lcsciences(houston,tx)平行合成技术合成,并且以混合物的形式在单一管中。表20:引物序列列表ω引物前体序列混合物首先使用taqhotstart2xmastermix(neb,ipswich,ma)进行扩增。pcr反应混合物组分如下表:表21:pcr混合物组分单位管1通用引物(comprm1和comprm2)nm(每种引物)500模板-ω引物前体混合物pm1taqhotstart2×mastermixx1总体积μl25热循环反应是在热循环仪dnaenginetetrad(来自bio-rad,hercules,ca)上进行。pcr温度程序如下:表22:pcr温度程序步骤温度(℃)时间活化1955分钟变性29530秒退火36030秒延伸46860秒转到2,10个循环5延伸67210分钟保持74永远使用实验i相同的琼脂糖凝胶电泳方法分析pcr产物。图20a泳道1显示为pcr的产物的琼脂糖凝胶电泳图像。可以看到如预期所示的95-130bp,中间为110bp分布的pcr产物。ω引物前体的pcr产物然后使用pcr纯化珠(agencourtampurexpsystem,来自beckmancoulter,brea,ca)进行纯化,纯化过程按照珠制造商说明进行。纯化的pcr产物的浓度使用agilent的bioanalyzer(santaclara,ca)进行检测。然后,pcr产物的du碱基使用udg/eda处理去除。udg消化溶液配制方法参照下表。udg和udg缓冲液购买自neb公司(ipswich,ma)。溶液在37℃温育60分钟。然后,取2μl200mmeda(来自于sigma-aldrich,st.louis,mo)加入溶液中在37℃温育另外60分钟。表23:反应混合物组分单位管1总体积μl10pcr产物μl8udg(5u/μl)μl1udg缓冲液,10×μl1接力pcr使用扩增的并且去除du的特异引物前体来进行,模板为人基因组dna,聚合酶选择phusionhotstartflex聚合酶mastermix(来自于neb,ipswich,ma)。各个反应成分的组分如下表。管1中的反应是没有加入特异引物前体的阴性对照反应,管2为接力pcr的实验组反应。表24:反应混合物组分单位管1管2特异性引物前体nm(所有)1.4prepprm1nm200200通用引物(comprm1,comprm2)nm(每种引物)500500模板-人类基因组dnafm22phusionhotstartflex2×mastermixx11总体积μl2525热循环反应是在热循环仪dnaenginetetrad(frombio-rad,hercules,ca)上进行。pcr温度程序如下:表25:接力pcr温度程序使用与实验i相同的琼脂糖凝胶电泳方法分析接力pcr产物。图20b显示为pcr的产物结果图片。l道是dna序列梯,表示相应标准参照物的大小(碱基对或bp)。泳道1是管1的阴性对照的结果,在阴性对照中没有观察到pcr产物(下部条带是由于未使用的引物)。泳道2加样有管2的pcr产物,可以看到如预期所示的295-399bp,中间为317bp的条带的分布。实施例vii---利用pct扩增制备的特异性引物的多重接力pcr我们进行了实验以实施利用pct扩增制备的特异性引物的多重接力pcr。这个实验的方法在本公开的图11的示例中已经有介绍。根据本公开的设计方法,该实验设计了捕获基因组组装版本grch37/hg19的人基因组中靶区域的ω引物。捕获的靶区域是属于apc1基因的外显子区域,该基因位于人的第5号染色体,捕获区域的起始位点为112,173,776,终止位点为112,173,955。下面的表格列出了该实验的寡核苷酸序列。制备引物prepprm1和prepprm2设计好用于pcr扩增特异引物模板。prepprm2序列中嵌入了限制酶识别位点gctcttc,用于限制核酸酶bspqi对pcr产物的限制性酶切。特异引物模板apc1_chr5_112173790_p1和apc1_chr5_112173944_p2包含5’和3’的侧翼区段,设计用于通过prepprm1和prepprm2进行pcr扩增。特异引物模板的中间部分设计ω引物。特异引物apc1_chr5_112173790_p1_no3pflank和apc1_chr5_112173944_p2_no3pflank拥有与apc1_chr5_112173790_p1和apc1_chr5_112173944_p2相同的ω引物设计,但是却没有3’侧翼区段。他们是激活特异引物,用作参考序列以与接力pcr反应中对靶区域进行捕获的特异引物模板产生的引物比较。comprm1和comprm2引物是设计的接力pcr的通用引物。下表的寡核苷酸序列由lcsciences(houston,tx)提供并使用传统的方法合成。表26:引物序列列表为了扩增特异引物模板,2个特异性引物模板等浓度混合在一起,pcr成分如下表所示。反应中使用到的聚合酶为hotstartphusion聚合酶(来自于neb,ipswich,ma)。表27:反应混合物组分单位管1制备引物(prepprm1,prepprm2)nm(每种引物)500模板(apc1_chr5_112173790_p1)fm20模板(apc1_chr5_112173944_p2)fm20phusionhotstartflex2×mastermixx1总体积μl25热循环反应是在热循环仪dnaenginetetrad(来自于bio-rad,hercules,ca)上进行。pcr温度程序如下:表28:pcr温度程序步骤温度(℃)时间活化1985分钟变性29815秒退火36030秒延伸47260秒转到2,1个循环5变性69815秒延伸77260秒转到6,19个循环8延伸97210分钟保持104永远使用与实验i相同的琼脂糖凝胶电泳方法分析pcr产物。图21a泳道1显示为pcr产物的凝胶图像。2个pcr产物的预期大小分别为153bp和141bp。胶带位置符合预期。pcr产物然后使用pcr纯化珠(agencourtampurexpsystem,来自beckmancoulter,brea,ca)进行纯化,纯化过程按照珠制造商说明进行。纯化的pcr产物的浓度使用nanodrop分光计测量(来自于nanodropproducts,wilmington,de)。然后,使用下表中所示的反应组分,限制酶消化pcr产物。限制酶bspqi以及10xcutsmart的缓冲液购买自neb(ipswich,ma)。消化反应在制造商推荐的温度和时间下进行:50℃,30分钟。消化产物然后使用pcr纯化珠(agencourtampurexpsystem,来自beckmancoulter,brea,ca)进行纯化,纯化过程按照珠制造商说明进行。纯化后的消化产物使用与实验i相同的琼脂糖凝胶电泳方法分析。图21b泳道2显示为产物的凝胶图像。两个消化产物的预期大小分别为132bp和120bp。胶带位置符合预期。fig.21b泳道1为限制酶消化前的原始pcr产物。表29:反应混合物组分使用下表所示的反应组分,限制酶消化产物然后进行λ外切核酸酶消化,以生成单链特异引物序列。λ外切核酸酶以及10x反应缓冲液购买自neb(ipswich,ma)。消化反应在酶制造商推荐的条件下进行:37℃,30分钟进行。消化产物然后使用pcr纯化珠(agencourtampurexpsystem,来自beckmancoulter,brea,ca)进行纯化,纯化过程按照珠制造商说明进行。我们称产物为酶制备特异引物。表30:反应混合物组分单位管1限制性酶消化产物(20ng/μl)μl810×λ外切核酸酶反应缓冲液μl1λ外切核酸酶(1000u/ml)μl1总体积μl10使用酶制备的特异引物进行接力pcr反应,模板为人基因组dna,聚合酶选用phusionhotstartflex聚合物mastermix(fromneb,ipswich,ma)。各个反应体系的反应组分如下表。管1反应为阳性对照,使用了参考特异引物apc1_chr5_112173790_p1_no3pflank和apc1_chr5_112173944_p2_no3pflank。管2反应为实验组使用酶制备的特异引物的反应。管3为阴性对照反应管,不加基因组dna。表31:反应混合物组分单位管1管2管3特异性引物(参考)nm(每种引物)1特异性引物(酶制备的)nm(每种引物)11通用引物(comprm1,comprm2)nm(每种引物)500500500模板-人类基因组dnafm11phusionhotstartflex2×mastermixx111总体积μl252525热循环反应是在热循环仪dnaenginetetrad(来自于bio-rad,hercules,ca)上进行。pcr温度程序如下:表32:接力pcr温度程序步骤温度(℃)时间活化1985分钟变性29815秒退火ω引物3652小时延伸146860秒延伸257260秒转到2,1个循环6变性79815秒起始lib引物退火86030秒延伸196860秒延伸2107260秒转到7,1个循环11变性129815秒延伸1136860秒延伸2147260秒转到12,27个循环15延伸167210分钟保持174永远使用与实验i相同的琼脂糖凝胶电泳方法分析接力pcr产物。图21c显示为pcr产物的图像。l泳道是dna序列梯,表示相应标准参照物的大小(碱基对或bp)。泳道1显示的是管1的阳性对照的结果,使用的是传统方法合成的参考特异引物,泳道2是实验组管2的结果,使用酶制备的特异引物。泳道1的阳性对照中和泳道2的实验中可以看到约300bp的相同产物大小。大小与预期大小312bp相符。泳道3为管3的为阴性对照结果,可以看到没有产物条带在300bp附近。本专利中,说特性是可选的,那就意指给权利要求提供足够的支持(例如.,35u.s.c.112和或者epc的83条以及84条),权利要求包括参考这个可选方法特性的封闭的,排除的或者否定的语言。排除语言尤其是从包括任何附加主题排除特定描述的特征。比如,说a可以是药物x,这是为了给权利要求提供支持,该权利要求明确地表明a只由x组成,或者a不包括除了x以外的任何其它药物。否定的语言明确地从权利要求范围排除可选的特性。例如,说元件a可以包括x,这种语言是用于给权利要求提供支持,权利要求明确地表明a不包括x。排除或否定属于的非限制性例子包括:“仅(只)”、“单独”、“包括(含),由…组成”、“基本上包括、基本上由…组成”、“单独”、“无(没有)”、“在缺少…的情况下(例如,相同类型的其它项、结构和/或功能)”、“不包括(排除)”、“不包括”、“不(没有)”、“不能(不可以)”、或者这种语言的任何组合和/或变化。同样,参考“一”或“所述”也可以用于支持单数和复数的事件,除非上下文说明了是其他的意思。例如,“狗”包括对一只狗,不多于一只狗,至少一只狗,一群狗等等的支持。使得术语表示单数的非限制性例子包括“单个(一)”、“一”、“单独”,“仅仅一”,“不多于一”等等。使得术语表示(潜在或实际的)复数的非限制性例子包括“至少一”、“一个或多个”、“一个以上”、“两个或多个”、“多重”、“多个”、“任何组合”、“任何变更”、“任何一个或多个”等。权利要求书或说明书在一组的一个或多个成员之间包含或者,如果一个,多于一个,或者所有的组成员存在于,被使用于或者与一个产品或者过程相关,就考虑其满足,除非上下文说明了是其他的意思。此处给出的范围,包括端点。此外,这是可以理解的,除非另有说明,否则从语境与本领域普通技术人员的理解,数值被表述成范围可以说在本发明不同实施例的这个范围的任何特定的值或者子范围,一直到范围的下限值的十分之一,除非上下文说明了是其他的意思。本专利申请书中引用的所有出版物和专利此处并入为参考文献,就好像每个单独的出版物或专利单独和特别地被标明并入为参考文献一样。任何出版物的引用是因为其公开在申请日之前,并且不应该解释为承认本发明由于现有技术的方面而没有资格早于这种公开的承认。尽管参考了实施例已经特别地表示和描述了本发明,本领域技术人员将理解这里可以进行形式和细节的各种变化,而不背离所附权利要求所包括的本发明的范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1