一种多倍体基因组survey的方法与流程
本发明属于分子生物学技术领域,具体涉及一种多倍体基因组survey的方法。
背景技术:
基因组survey是指在没有参考基因组对物种基因组特征进行评估的情况下,利用二代测序技术,通过统计k-mer频率,有效的评估基因组大小、杂合度、重复序列比例等信息,为后续基因组denovo组装(从头组装)提供参考。现在的大型基因组测序普遍使用全基因组鸟枪法(whole-genome-shotgun,wgs),其中不乏高重复和高杂合的基因组。这类基因组将显著增加基因组组装的难度,导致结果的不完整和碎片化,并且仅根据测序得到的重复序列信息无法准确推算该物种基因组大小。此外,当基因组有极高的重复序列和杂合序列比例时,或该物种为多倍体物种时,直接使用当下的组装算法很难得到理想的组装结果。因此在基因组测序前,对物种基因组特征进行准确评估以便制定合适的测序和组装方案十分重要。估算物种基因组大小通常有三方法:一是用流式细胞仪检测细胞核内dna的总量;二是用核型分析的方法,在显微镜下识别中期染色体数量、倍性以及大小,实现染色体长度和染色体大小的相对定量;三是基于k-mer分析,通过二代基因组测序估算基因组大小等信息。k-mer分析因其成本低,难度低,能得到更多分析结果,已成为目前获取基因组信息使用最多的技术手段。
随着高通量测序技术的快速发展和测序成本的迅速降低,基因组测序的需求不断增加。为了应对越来越多的多倍体物种测序的需求,我们需要对更多更复杂的多倍体物种基因组特征进行评估。但是目前的基因组特征评估方法仅对二倍体物种有较高准确度,而多倍体物种的基因组评估结果与真实结果往往有很大差异,因此急需一种可靠的多倍体基因组survey方法。
技术实现要素:
为了克服目前基因组特征评估方法对多倍体物种评估准确率低的问题,本发明提供了一种多倍体基因组survey方法,能准确评估多倍体物种的基因组特征,提高评估准确率,其具体技术方案如下:
一种多倍体基因组survey的方法,包含如下步骤:
步骤1,基因组dna提取与测序:
(1)选取生物个体的肌肉组织,切取5-20mg组织到2ml离心管中,用手术剪剪碎,再用匀浆机打碎;
(2)加入400ulacl溶液和20ul的蛋白酶k;
(3)震荡1~2min使之混匀,然后置于55℃放置3h,在此期间每半小时取出混匀,使之充分裂解,裂解完全的样品为澄清透明;
(4)取出样品,待降至室温后轻轻震荡均匀;
(5)在经过处理好的样品中,依次加入300ulext溶液和300ulab溶液,用力摇匀后进行离心处理,使溶液分层,上层为蓝色的抽提层,下层为透明水相,两层中间会有部分沉淀层,dna在下层水相中;
(6)将枪头穿过上层溶液,深入到下层溶液,将下层溶液仔细吸出到吸附柱中,尽量避免吸到上层溶液和中间层的沉淀;
(7)将吸附柱放入离心机,进行离心处理,然后取下吸附柱,倒掉收集管中的废液;
(8)将吸附柱放回收集管中,加入500ul漂洗液,进行离心处理;
(9)重复步骤(8)一次;
(10)取下吸附柱,弃去收集管中的废液,将吸附柱放回收集管中,进行离心处理,以除去残留漂洗液;
(11)将柱放入新的洁净离心管中,在柱中央加入50~100ul洗脱缓冲液,室温放置2~3min,然后进行离心处理,离心管中的液体即为基因组dna,样品置于-4℃或-20℃保存;
(12)利用nanodrop-1000浓度测试仪准确测定样本dna浓度和od比值(a260/a280、a260/a230);要求样品dna浓度在100ng/ul以上,样品dna体积至少60ul,a260/a280值在1.8~2.0,a260/a230值在2.0~2.4;
(13)质控合格的样品使用illunimahiseq2000平台进行高通量测序;
步骤2,基因组测序产出数据质量控制:
基于多倍体基因组二代高通量测序数据的质量控制:使用fastqc软件对测序数据质量进行评估,保证测序数据符合后续的分析需求;
使用trimmomatic软件过滤低质量的reads,即任何一端含有接头的双端reads;单端reads中未测出碱基超过该reads长度10%的双端reads;单端reads中低质量碱基数超过该reads碱基数50%的双端reads;
随机抽取10000对双端reads用blast软件与nt数据库(nucleotidesequencedatabase)比对,确保测序的样品没有明显污染;
步骤3,多倍体基因组特征评估:
基于高质量的多倍体物种基因组测序数据的k-mer分析:利用步骤2质量控制方法获得的高质量测序数据,用k=17参数计算k-mer数量和k-mer频率分布情况,公式
同时将k小于2的情况认为是错误,用公式grevise=g×(1-errorrate)对基因组大小进行校正,其中,grevise:矫正后基因组大小;errorrate:错误率;
用公式
步骤4,多倍体基因组倍性变化分析:
基于渐渗模型的基因组倍性变化分析:利用渐渗模型公式
所述步骤1(5)中,所述离心参数设置为12000rmp,离心5~6min;
所述步骤1(7)中,所述离心参数设置为8000rmp离心1~2min;
所述步骤1(8)中,所述离心参数设置为8000rmp离心1~2min;
所述步骤1(10)中,所述离心参数设置为12000rmp,室温离心1~2min;
所述步骤1(11)中,所述离心参数设置为12000rmp,室温离心1~2min。
本发明的一种多倍体基因组survey方法,与现有技术相比,有益效果为:
一、本发明方法设计的时已考虑到了多倍体物种基因组序列之间复杂关系,专门设计了针对多倍体物种基因组大小、杂合率的分析方法,能有效解决现有方法评估多倍体基因组评估准确率低的问题;
二、本发明创造性地提出并构建了多倍体基因组的渐渗模型。基于所述步骤4中,渐渗模型的基因组被性分析可以计算多倍体基因组同源率和二倍化率,填补了现有分析方法中这一结果的空缺
三、本方法理论上可以对任何真核多倍体基因组进行survey分析,因而本方法可以在多倍体基因组评估上得到广泛应用,为多倍体基因组大小、杂合率、同源率评估等应用提供了准确有效的分析方法。
四、与现有技术相比,本方法提高了90%的评估准确率。
附图说明
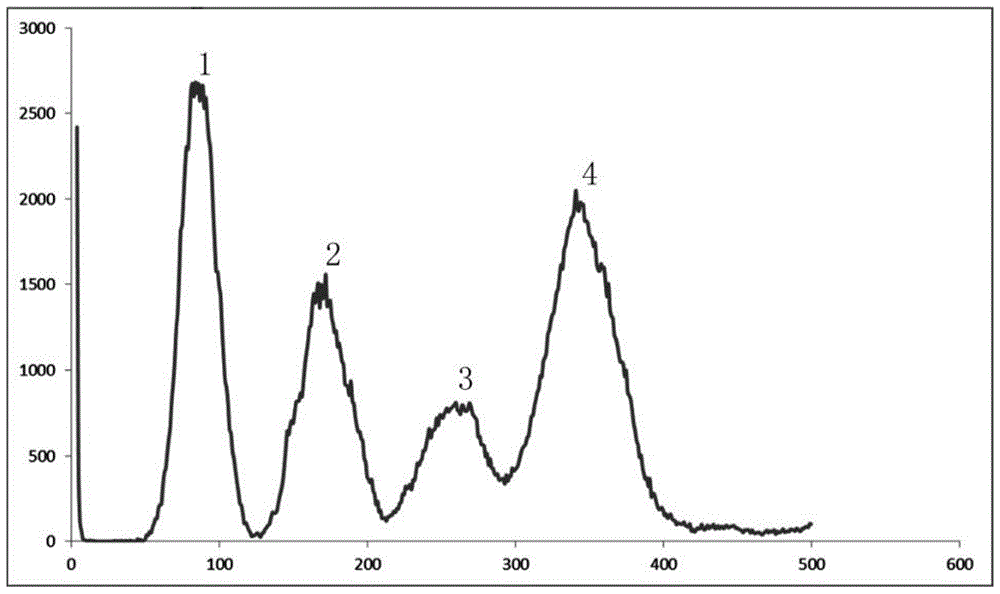
图1为本发明实施例1的四倍体基因组k-mer频率分布示意图:其中,1-全基因组杂合峰,2-二倍体主峰,3-四倍体杂合峰,四倍体主峰;
图2为本发明实施例1的四倍体基因渐渗过程示意图:其中,a-祖先染色体,a’-渐渗过程发生的染色体,b-渐渗过程结束的染色体;
图3为本发明实施例1的四倍体基因渐渗模型结构示意图。
具体实施方试
下面结合具体实施案例和附图1-3对本发明作进一步说明,但本发明并不局限于这些实施例。
实施例采用四倍体泥鳅基因组,选取一条合适的泥鳅个体,取其肌肉进行基因组dna提取,提取方法为上海捷瑞生物工程有限公司的细胞/组织基因组dna提取试剂盒(gk0122)。
实施例1
一种多倍体基因组survey的方法,具体包含如下步骤:
步骤1,基因组dna提取与测序:
(1)切取10mg组织到2ml离心管中,用手术剪(酒精消毒)剪碎,再用匀浆机打碎;
(2)加入400ulacl溶液和20ul的蛋白酶k;
(3)震荡混匀1分钟,然后置于55℃放置3h,在此期间每半小时取出混匀,有助于充分裂解,裂解完全的的样品应澄清透明;
(4)取出样品,待降至室温时轻轻震荡均匀;
(5)在经过处理好的样品中,依次加入300ulext溶液和300ulab溶液,用力摇匀,然后12000rmp离心5min。溶液将分层,上层为蓝色的抽提层,下层为透明水相,两层中间可能会有部分沉淀层,dna在下层水相中;
(6)将枪头穿过上层溶液,深入到下层溶液,将下层溶液仔细吸出到吸附柱中,尽量避免吸到上层溶液和中间层的沉淀;
(7)8000rmp离心1min,取下吸附柱,倒掉收集管中的废液;
(8)将吸附柱放回收集管中,加入500ul漂洗液,8000rmp室温离心1min;
(9)重复步骤(8)一次;
(10)取下吸附柱,弃去收集管中的废液。将吸附柱放回收集管中,12000rmp,室温离心1min,以除去残留漂洗液;
(11)将柱放入新的洁净1.5ml离心管中,在柱中央加入80ul洗脱缓冲液,室温放置2min,然后12000rmp,室温离心1min。离心管中的液体即为基因组dna,样品可于-4℃或-20℃保存;
(12)利用nanodrop-1000浓度测试仪准确测定样本dna浓度和od比值(a260/a280、a260/a230);要求样品dna浓度在100ng/ul以上,样品dna体积至少60ul,a260/a280值在1.8,a260/a230值在2.4;
(13)质控合格的样品使用illunimahiseq2000平台进行高通量测序;
步骤2,基因组测序产出数据质量控制:
高通量测序数据质量控制的具体步骤如下:
(1)获取原始的测序数据,使用fastqc软件对原始数据质量进行评估,确保数据满足二代测序数据的分析要求;
(2)使用trimmomatic软件对原始数据按照下列规则进行过滤:过滤掉任何一端含有接头序列的双端reads;当测序的单端read中含有未测出碱基(n)超过该条read长度的10%时,过滤这对双端reads;当单条测序reads中含有的低质量(小于等于5)碱基数超过该条reads长度的50%时,过滤这对双端reads,使用参数:-minlen50-slinwin4-slinqsum15-heading3-trailing3;
(3)随机抽取10,000对reads,使用blastn工具将reads比对到nt数据库(nucleotidesequencedatabase)数据库,确保比对次数最多序列是泥鳅的近缘物种,以排除样品污染的可能;
(4)经过质量控制,成功得到37gb高质量测序数据;
步骤3,多倍体基因组特征评估:
利用经过质控的高质量测序数据进行基因组特征评估,具体方法如下:
(1)使用k-mer分析软件gce计算2.4步骤得到的数据,具体参数如下:-m1-d8-b0-h1,计算得到250,217,368,293条k-mer和k-mer的分布频率,以及基因组gc含量40.98%;
(2)如图1所示,根据四倍体基因组的k-mer分布特征,结合k-mer分布频率,可以算得多倍体主峰对应的k-mer深度为92;
(3)使用基因组大小计算公式
(4)用误差矫正公式grevise=g×(1-errorrate)可以算得矫正后四倍体泥鳅基因组大小为2,632.9mb;
(5)根据k-mer分布频率和杂合率估算公式
步骤4,多倍体基因组倍性变化分析:
通过逐步区域替换的方法模拟四倍体基因组二倍化,在替换基础上引入随机突变,模拟杂合率为1%,以构建四倍体基因组二倍化的渐渗模型,如图2-3所示,根据渐渗模型公式
实施例2
一种多倍体基因组survey的方法,具体包含如下步骤:
步骤1,基因组dna提取与测序:
(1)切取7mg组织到2ml离心管中,用手术剪(酒精消毒)剪碎,再用匀浆机打碎;
(2)加入400ulacl溶液和20ul的蛋白酶k;
(3)震荡混匀1分钟,然后置于55℃放置3h,在此期间每半小时取出混匀,有助于充分裂解,裂解完全的的样品应澄清透明;
(4)取出样品,待降至室温时轻轻震荡均匀;
(5)在经过处理好的样品中,依次加入300ulext溶液和300ulab溶液,用力摇匀,然后12000rmp离心5min。溶液将分层,上层为蓝色的抽提层,下层为透明水相,两层中间可能会有部分沉淀层,dna在下层水相中;
(6)将枪头穿过上层溶液,深入到下层溶液,将下层溶液仔细吸出到吸附柱中,尽量避免吸到上层溶液和中间层的沉淀;
(7)8000rmp离心1min,取下吸附柱,倒掉收集管中的废液;
(8)将吸附柱放回收集管中,加入500ul漂洗液,8000rmp室温离心1min;
(9)重复步骤(8)一次;
(10)取下吸附柱,弃去收集管中的废液。将吸附柱放回收集管中,12000rmp,室温离心1min,以除去残留漂洗液;
(11)将柱放入新的洁净1.5ml离心管中,在柱中央加入50ul洗脱缓冲液,室温放置2min,然后12000rmp,室温离心1min。离心管中的液体即为基因组dna,样品可于-4℃或-20℃保存;
(12)利用nanodrop-1000浓度测试仪准确测定样本dna浓度和od比值(a260/a280、a260/a230);要求样品dna浓度在100ng/ul以上,样品dna体积至少60ul,a260/a280值在1.9,a260/a230值在2.3;
(13)质控合格的样品使用illunimahiseq2000平台进行高通量测序;
步骤2,基因组测序产出数据质量控制:
高通量测序数据质量控制的具体步骤如下:
(1)获取原始的测序数据,使用fastqc软件对原始数据质量进行评估,确保数据满足二代测序数据的分析要求;
(2)使用trimmomatic软件对原始数据按照下列规则进行过滤:过滤掉任何一端含有接头序列的双端reads;当测序的单端read中含有未测出碱基(n)超过该条read长度的10%时,过滤这对双端reads;当单条测序reads中含有的低质量(小于等于5)碱基数超过该条reads长度的50%时,过滤这对双端reads。使用参数:-minlen50-slinwin4-slinqsum15-heading3-trailing3;
(3)随机抽取10,000对reads,使用blastn工具将reads比对到nt数据库(nucleotidesequencedatabase)数据库,确保比对次数最多序列是泥鳅的近缘物种,以排除样品污染的可能;
(4)经过质量控制,成功得到37gb高质量测序数据;
步骤3,多倍体基因组特征评估:
利用经过质控的高质量测序数据进行基因组特征评估,具体方法如下:
(1)使用k-mer分析软件gce计算2.4步骤得到的数据,具体参数如下:-m1-d8-b0-h1,计算得到255,192,472,392条k-mer和k-mer的分布频率,以及基因组gc含量40.53%;
(2)根据四倍体基因组的k-mer分布特征,结合k-mer分布频率,可以算得多倍体主峰对应的k-mer深度为93;
(3)使用基因组大小计算公式
(4)用误差矫正公式grevise=g×(1-errorrate)可以算得矫正后四倍体泥鳅基因组大小为2653.3mb;
(5)根据k-mer分布频率和杂合率估算公式
步骤4,多倍体基因组倍性变化分析:
通过逐步区域替换的方法模拟四倍体基因组二倍化,在替换基础上引入随机突变,模拟杂合率为1%,以构建四倍体基因组二倍化的渐渗模型,根据渐渗模型公式
实施例3
一种多倍体基因组survey的方法,具体包含如下步骤:
步骤1,基因组dna提取与测序:
(1)切取10mg组织到2ml离心管中,用手术剪(酒精消毒)剪碎,再用匀浆机打碎;
(2)加入400ulacl溶液和20ul的蛋白酶k;
(3)震荡混匀2分钟,然后置于55℃放置3h,在此期间每半小时取出混匀,有助于充分裂解,裂解完全的的样品应澄清透明;
(4)取出样品,待降至室温时轻轻震荡均匀;
(5)在经过处理好的样品中,依次加入300ulext溶液和300ulab溶液,用力摇匀,然后12000rmp离心6min。溶液将分层,上层为蓝色的抽提层,下层为透明水相,两层中间可能会有部分沉淀层,dna在下层水相中;
(6)将枪头穿过上层溶液,深入到下层溶液,将下层溶液仔细吸出到吸附柱中,尽量避免吸到上层溶液和中间层的沉淀;
(7)8000rmp离心2min,取下吸附柱,倒掉收集管中的废液;
(8)将吸附柱放回收集管中,加入500ul漂洗液,8000rmp室温离心2min;
(9)重复步骤(8)一次;
(10)取下吸附柱,弃去收集管中的废液。将吸附柱放回收集管中,12000rmp,室温离心2min,以除去残留漂洗液;
(11)将柱放入新的洁净1.5ml离心管中,在柱中央加入100ul洗脱缓冲液,室温放置3min,然后12000rmp,室温离心2min。离心管中的液体即为基因组dna,样品可于-4℃或-20℃保存;
(12)利用nanodrop-1000浓度测试仪准确测定样本dna浓度和od比值(a260/a280、a260/a230);要求样品dna浓度在100ng/ul以上,样品dna体积至少60ul,a260/a280值在2.0,a260/a230值在2.0;
(13)质控合格的样品使用illunimahiseq2000平台进行高通量测序;
步骤2,基因组测序产出数据质量控制:
高通量测序数据质量控制的具体步骤如下:
(1)获取原始的测序数据,使用fastqc软件对原始数据质量进行评估,确保数据满足二代测序数据的分析要求;
(2)使用trimmomatic软件对原始数据按照下列规则进行过滤:过滤掉任何一端含有接头序列的双端reads;当测序的单端read中含有未测出碱基(n)超过该条read长度的10%时,过滤这对双端reads;当单条测序reads中含有的低质量(小于等于5)碱基数超过该条reads长度的50%时,过滤这对双端reads。使用参数:-minlen50-slinwin4-slinqsum15-heading3-trailing3;
(3)随机抽取10,000对reads,使用blastn工具将reads比对到nt数据库(nucleotidesequencedatabase)数据库,确保比对次数最多序列是泥鳅的近缘物种,以排除样品污染的可能;
(4)经过质量控制,成功得到37gb高质量测序数据;
步骤3,多倍体基因组特征评估:
利用经过质控的高质量测序数据进行基因组特征评估,具体方法如下:
(1)使用k-mer分析软件gce计算2.4步骤得到的数据,具体参数如下:-m1-d8-b0-h1,计算得到253,936,781,425条k-mer和k-mer的分布频率,以及基因组gc含量40.75%;
(2)根据四倍体基因组的k-mer分布特征,结合k-mer分布频率,可以算得多倍体主峰对应的k-mer深度为93;
(3)使用基因组大小计算公式
(4)用误差矫正公式grevise=g×(1-errorrate)可以算得矫正后四倍体泥鳅基因组大小为2639.7mb;
(5)根据k-mer分布频率和杂合率估算公式
步骤4,多倍体基因组倍性变化分析:
通过逐步区域替换的方法模拟四倍体基因组二倍化,在替换基础上引入随机突变,模拟杂合率为1%,以构建四倍体基因组二倍化的渐渗模型,根据渐渗模型公式
本发明的实施例,以上结果均符合预期,与现有技术相比,本方法评估准确率提高了90%。
- 还没有人留言评论。精彩留言会获得点赞!